Rasha Mahdi Abdulkader![]() | Atyaf Ismaeel Altameemi
| Atyaf Ismaeel Altameemi![]() | Rasha Basim Issa
| Rasha Basim Issa![]() | ShaymaaTaha Ahmed
| ShaymaaTaha Ahmed![]() | Qusay Kanaan Kadhim
| Qusay Kanaan Kadhim![]() | Alyaa Aziz Ahmed
| Alyaa Aziz Ahmed![]() | Wasan Abdulwahab Faiq Alsiadi*
| Wasan Abdulwahab Faiq Alsiadi*![]()
© 2026 The authors. This article is published by IIETA and is licensed under the CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
OPEN ACCESS
Diabetic vascular aging contributes substantially to cardiovascular complications in diabetes, yet the molecular drivers underlying this process remain incompletely characterized. This study presents a weighted Hyperlink-Induced Topic Search (HITS) framework for prioritizing candidate hub genes by integrating diabetes-associated genome-wide association study (GWAS) signals, differentially expressed genes from GSE5281 and GSE33000, and a human protein–protein interaction (PPI) network. Biological evidence and network-topological information were incorporated into weighted edges, after which hub and authority scores were iteratively estimated to rank genes. Differential expression analysis was performed after quality control, normalization, probe annotation, and missing-value imputation. The final ranking was examined across alternative weighting settings, with α = 0.5 selected to balance biological and topological contributions. The algorithm showed stable convergence within approximately 500 iterations and prioritized CCR2, VCAM1, CSF1R, ITGAM, and MODY1 as candidate hub genes. Functional enrichment analysis associated the ranked genes with immune-response regulation, membrane trafficking, lipid metabolism, neutrophil degranulation, and osteoclast-differentiation pathways. The framework provides a network-based strategy for integrating heterogeneous biological evidence in gene prioritization. This approach may support hypothesis generation for mechanism-focused follow-up studies. Because the analysis relies on public datasets and inferred interaction networks, the reported genes should be regarded as candidates requiring validation in independent patient cohorts and experimental models of diabetic vascular aging.
diabetic vascular aging, candidate hub genes, weighted Hyperlink-Induced Topic Search algorithm, protein–protein interaction network, genome-wide association study, differential gene expression, network-based bioinformatics
Computational methods for robust gene prioritization in complex diseases like diabetes remain a significant challenge in bioinformatics. From an information systems perspective, the proposed weighted Hyperlink-Induced Topic Search (HITS) framework contributes to computational network intelligence by integrating graph-based ranking, biological weighting, and topological analysis into a scalable decision-support model for biological knowledge discovery [1]. Unlike traditional bioinformatics pipelines, the proposed method emphasizes efficient information propagation and ranking in complex heterogeneous networks. This study addresses this gap by introducing a weighted HITS algorithm, integrating both network topology and biological information. The biological motivation stems from the critical need to understand vascular aging in diabetes, a process accelerated by Vascular Smooth Muscle Cell (VSMC) senescence [2]. The International Diabetes Federation's Diabetes Atlas, 10th edition, states that the prevalence of diabetes in those between the ages of 20 and 80 worldwide was 11.5% in 2021 (537.7 million people), and it is predicted for increase to 13.3% (793.4 million people) by 2050 [3]. Microvascular problems, such diabetic retinopathy and nephropathy, as well as macrovascular issues including coronary artery disease, diabetic cardiomyopathy, etc., are the two categories of CVD complications linked to diabetes [4]. Vascular aging is the term used to describe the circulatory system's structural and functional degenerative alterations. These alterations impact the prognosis, severity, progression, and threshold of a number of age-related disorders [5]. Vascular aging is a major contributor to diabetes-related cardiovascular disease and is highly prevalent among diabetic patients [6]. One significant vascular aging characteristic is vascular calcification [7]. A prevalent macro vascular consequence Refractory hypertension, atherosclerosis, amputation, stroke, and kidney failure are all made more likely in diabetic individuals due to arterial calcification, which also increases the risk of vascular events and death. One of the main cell types that comprise the vascular wall is Vascular Smooth Muscle Cells (VSMCs), have the ability to contract and relax to regulate blood channel width and flow [8].
Diabetes has a significant impact on VSMC senescence. VSMC senescence is accelerated in diabetes because to the hyperglycemic condition, which raises inflammation and intracellular oxidative stress and damages VSMC proteins and DNA [9]. Conversely, VSMC senescence causes the arterial wall to become thicker and less elastic, which can result in atherosclerotic plaque formation and vascular sclerosis, raising the risk of cardiovascular disease [10]. Thus, VSMC senescence prevention and treatment may assist diabetic patients reduce their risk of cardiovascular issues [11].
A typical method for analyzing gene and protein expression profiles is bioinformatics, which makes it easier to identify the molecular pathways driving particular clinical alterations [12]. Using the bioinformatics approach, this work investigated the molecular processes and related genes that cause diabetic vascular aging with reference to pathogenic mechanism from diabetic vascular aging caused by VSMC senescence.
The Protein-Protein Interaction (PPI) is essential to every biological function and is frequently dysregulated in illnesses[13].While many proteins interact with many partners, a sizable fraction of proteins only interact with a small number of partners[14].These interactions are important for signal transmission, apoptosis, cell development, and other biological processes that support tissue regeneration, cell proliferation, and control[15].With the existence of particular domains, the majority of proteins are members of protein families that are related in evolution. High accuracy is known to occur when certain proteins from a family of proteins interact with one another. Analyzing the specificity of these interactions becomes considerably more difficult in this situation [16].
The PPIs are now probably going to be one of the next big therapeutic target classes. The majority of transcription factors (TFs), which are known to primarily function through these interactions, are now the most appealing area of the therapeutic development process [17]. Integrating structural data into useful information for the drug discovery process is a significant challenge in this field. Multiprotein complexes and PPIs are crucial components of every living thing's biological processes [18]. Finding the roles of other interacting proteins can help uncover the functions of an unknown protein in addition to these cellular processes and signal transductions. A process can start with a protein that is found in the nucleus (such transcription factors) and finish with the membrane proteins since cellular pathways can be so extensive. Conversely, there are tiny pathways with a limited number of proteins. While some proteins—like PI3KCA, STAT3, and AKT1are engaged in multiple cellular pathways, others—like GLUT4, which is specialized in glucose transporter made to carry out a single task [19]. Interactions between proteins within a cell are inevitable. The entire pathway may be impacted by a change in one protein because the proteins are interconnected [20].
It was first introduced by Kleinberg in 1998, and is known as HITS in the field of web structure mining [21]. Prior to joining them in the link structure, Kleinberg separated authority pages and hub pages from network pages [22]. The former offers the best information on search-related issues; the more network pages cite it, the greater its authority value [23]. The latter offers significant linkages and the more authoritative pages it cites, the greater its hub value. Web searches frequently use the HITS algorithm, which effectively addresses various real-world including web community [24]. An innovative computational technique is presented in this study to identify essential proteins using weighted PPI networks and the HITS algorithm. First, we create a directed network from the original undirected PPI network. After that, we weighted PPI networks using biological data and network topological properties and assess three factors protein functions false positives and false negatives, and protein locations [25].
The biological information employed in this technique includes data on gene expression, and Gene Ontology (GO) annotation [26]. As an illustration understanding PPI networks' topological characteristics. Then, using the authority and hub principles produced using HITS method, the rank the proteins [27].
Recent advances in computational network analysis have enabled efficient identification of influential nodes in complex biological systems. Algorithms such as PageRank, DeepWalk, and graph-based centrality measures have been widely applied for protein prioritization. However, many existing methods focus mainly on network topology without integrating biological weighting mechanisms. Therefore, this study proposes a weighted HITS framework that combines topological structure with biological annotations for improved hub-gene discovery.
This research used protein-protein interactions to discover the hub genes responsible for diabetes and HITS algorithm. Finally, we obtained the top 5 genes related to diabetes. A critical aspect of this study is the introduction of a novel computational method to determine important proteins via a weighted PPI network and the HITS algorithm. This advanced bioinformatics approach, which utilizes principles central to machine learning and network analysis, was essential for analyzing complex biological data and network topology. The successful application of the HITS algorithm allowed for the reliable identification of essential hub genes (CCR2, VCAM1, CSF1R, ITGAM, and MODY1) strongly linked to diabetes and vascular aging. This extensive bioinformatics investigation deepens our understanding of the molecular basis of diabetes and may provide a crucial foundation for developing new therapeutic drugs for diabetic patients. Recent advances in computational network analysis have enabled efficient identification of influential nodes in complex biological systems. Algorithms such as PageRank, DeepWalk, and graph-based centrality measures have been widely applied for protein prioritization. However, many existing methods focus mainly on network topology without integrating biological weighting mechanisms.
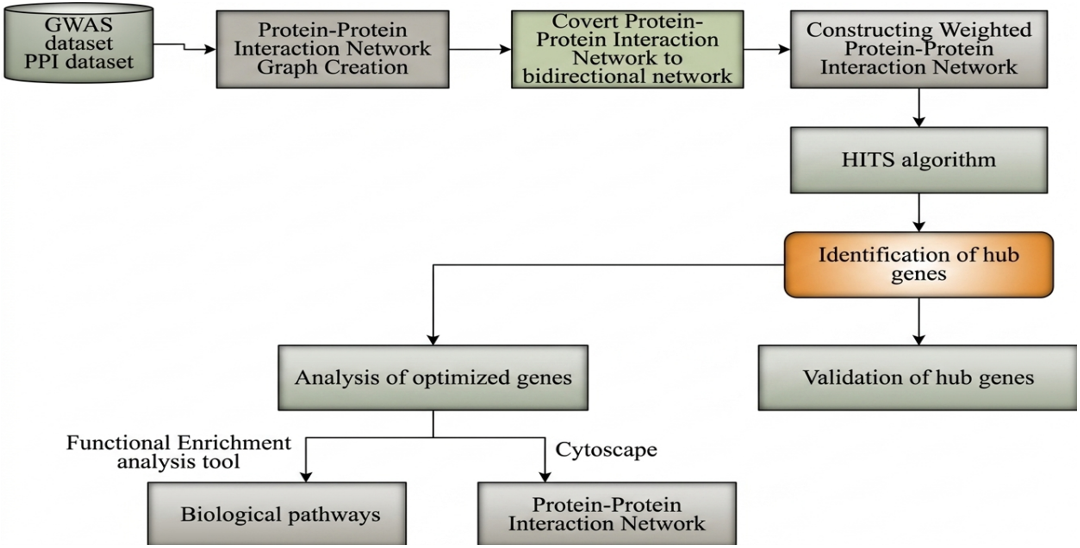
A proposal used the dataset of PPI network of human is integrated from the Human Protein Reference Database (HPRD), has 46496 genes. The datasets Genome-Wide Association Study (GWAS) the GWAS association results for diabetes were downloaded. GWAS has760 genes involved in diabetes. Methods of detailed description of all the algorithms, formulas and methods that helped to present proposal used the HITS algorithm and extra-biological information to identify and evaluate hub genes, as shown in Figure 1. The proposed framework consists of the following sequential steps:
Step 1: Construction of the weighted PPI network.
Step 2: Integration of biological information obtained from GWAS and Differentially Expressed Genes (DEGs) datasets.
Step 3: Application of the weighted HITS algorithm to calculate hub and authority scores.
Step 4: Adjustment of the α parameter to balance biological and topological influences.
Step 5: Identification and evaluation of hub genes using functional enrichment analysis.
Building a Weighted Network of Protein-Protein Interactions, the most common way to depict a protein-protein interaction network is as a directionless graph D = (V, P), where V is a customary of vertices that stands for proteins and P is a collection of all interactions between proteins [28].
We started from the assumption that protein connections were interactive and transform the directionless PPI network D = (V, P) into a bidirectional network D'= (V, P') was similar in order to defy accepted wisdom. Mentioning certain networks in biology, like kinase networks, are exempt from the mathematical process that converts an undirected graph into a directed graph. The high number of false positives in high through put PPI networks, and false negatives will affect the prediction accuracy. In order to resolve this issue, we weigh edges separately using network topological traits and biological information. Nodes with high-quality biological data are anticipated to be directed to using nodes that indicate higher topological data that vice versa, based on the HITS algorithm. The building of the weighted PPI network is illustrated with an example in Figure 1.
Figure 1. Proposed method
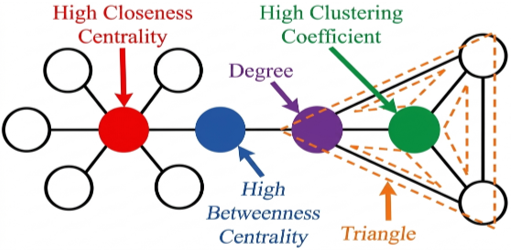
Weighted edge in network topology: Edge Betweenness Centrality (EBC) is a way to quantify a node's impact on a graph's information flow. Finding nodes that link two different areas of a network is a common usage for it. A measure of centrality that goes beyond betweenness is the number of times node is seen situated along the shortest path between two other nodes. It is equal to the sum of the shortest paths between each vertex and every other vertex that go via that node. If we assume that the shortest paths are used for item transfer, the transmission of items through the network is significantly impacted by a node with high betweenness centrality. A quickest path is taken by the brain to digest information in order to conserve time and energy, according to studies. In order to reflect the shortest paths in our model, we chose proximity and betweenness centrality as measurements. Betweenness Centrality CB for the graph D = (V, P) (p) is given in Eq. (1). EBC (p) can be defined, as follows:
$\operatorname{EBC}(\mathrm{p})=\sum \mathrm{s} \neq \mathrm{p} \neq \mathrm{t} \neq$ (1)
It showed the total number of shortest routes between nodes s and t by σst, while the numeral of pathways that pass via p is represented by σst (p), as shown in Figure 2.
Biological data weighted edge: Several genes linked to diabetes were found via Genome-Wide Association Studies (GWASs). The biological pathway is a method using which a gene's information is utilized in a creation from a useful gene produce.
Figure 2. Betweenness centrality [29]
2.1 Data collection and preprocessing
The human PPI network was integrated from HPRD. Genes associated with diabetes were obtained from a GWAS dataset. To ensure the robustness and reliability of our bioinformatics analysis, the raw gene expression data from the integrated datasets (GSE5281 and GSE33000) underwent a comprehensive preprocessing pipeline. Initial quality control was performed using the ArrayQualityMetrics package in R to identify and exclude outlier arrays, resulting in a final network of 5,664 nodes and 37,219 edges for analysis. To reduce the impact of background noise and normalization, the Robust Multi-Array Average (RMA) approach was used. technical noise and ensure sample comparability. Probe sets were then annotated using the platform-specific annotation files to convert gene symbols. The arithmetic mean was used to sum up the expression value for genes represented by many probe sets. Missing values were imputed using the k-nearest neighbors (KNN) algorithm (with k = 10) to preserve statistical power. Differential expression analysis between diabetic vascular aging samples and controls was conducted using the Lima package in R, applying a false discovery rate (FDR) correction of < 0.05 and an absolute log2 fold change threshold of > 1 to identify statistically significant DEGs. The final list of DEGs was mapped onto the human PPI network retrieved from the HPRD, retaining only interactions where both partner proteins corresponded to genes in our filtered DEG list, thus constructing a high-confidence, context-specific PPI network for subsequent analysis. We clarified that, as the HITS methodology is an unsupervised network ranking technique, traditional train/test splitting was not performed.
The PPI network of human is integrated from HPRD, and had 3379 genes. The datasets GWAS retrieved the diabetes GWAS association findings from (http://jjwanglab.org/ gwasdb). The GWAS has 769 genes involved in diabetes. Details of the used datasets are presented in Table 1.
Table 1. Brief description of the datasets
|
Dataset |
|
|
Homo sapiens PPI network |
5664 nodes and 37219 interactions |
|
GWAS |
769 diabetes-associated genes |
2.2 Hyperlink Induced Topic Search algorithm
The HITS algorithm an iterative algorithm was first suggested to evaluate a significance of online pages [30]. The HITS algorithm rates the web page via analyzing all of its in-links and out-links, which is reliant on the search query. Each page receives two attributes in the HITS algorithm: the hub and the authority. Many high-quality hub pages will link to the authority, which is a high-quality authority page. The values of each page's authority that the page hub links to together make up its value [31]. Hub is a top-notch hub page that links to numerous top-notch authority pages. The total hub values that point to the page make up the page authority value.
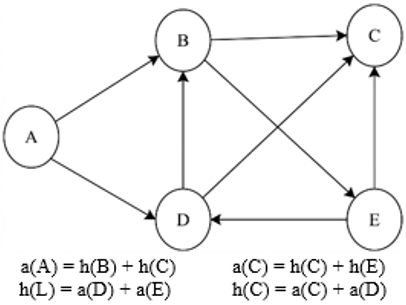
Demonstrates how to calculate the importance of the authority and hub, as seen in Figure 3.
Figure 3. An example of how to calculate hub and authority values simply
The Hub (h) score and Authority (a) score for a node is calculated the technique below:
The authority and hub scores of page p are denoted by a (p) and h (p), respectively. The set of referrer and reference pages of page p are indicated by a(p) and h(p), respectively. There are multiple steps in the HITS algorithm:
Calculate a (p) and h (p) in a manner that reinforces each other (see Eqs. (2) and (3)).
$ a(p)=\sum_{q \in B(p)} h(q)$ (2)
$ h(p)=\sum_{q \in F(p)} a(q)$ (3)
To normalize it, divide each web page's authority by the highest authority (see Eq. (4)).
$ a(p)=\frac{a(p)}{\max (a(p))}$ (4)
To normalize, divide each web page's hub by the highest hub it as shown in Eq. (5).
$ h(p)=\frac{h(p)}{\max (h(p))}$ (5)
Repeat step 2 until the weight difference between the previous iteration and the current one is zero after the system has reached a steady state with a convergence between u and h. iteration, with the current iteration being below the specified limit.
2.3 Protein–Protein Interaction network
Understanding PPIs is critical for comprehending cell physiology in both normal and diseased states since they allow one to observe the interactions between proteins, which are necessary for nearly every process in a cell [32]. Given that medications might impact PPIs; it is also crucial in the development of new therapies. Data integrated diabetes database that integrates high-throughput omics data, such as the results of GWAS, provided the PPI details for the relevant genes [33].
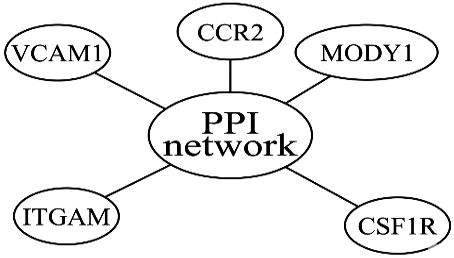
The PPI visual network was constructed using the program Cytoscape.Finally, using the "CytoHubba" plug-in in Cytoscape, the top ten genes from the interesting genes with the strongest relationship were selected as the hub genes for the network. According to Figure 4.
Figure 4. Network of Protein-Protein Interactions [34]
2.4 Identifying hub gene based on Hyperlink-Induced Topic Search algorithm
The suggested HITS approach is applied, and weighted PPI networks are constructed. The ultimate score is calculated by combining the hub value. A notice that in every change in weight some common genes appear and new genes also appear, this led to conclusion that it is better to make the value α half to each of the h = 0.5 and a = 0.5 to make the network balanced, as presented in the Table 1, to fully assess each gene's significance, where [0, 1] utilized to modify a ratio (α) between these two marks.
Only for a value of the topological data determines the sorting score, which is 0. A sorting score is calculated using a biological data if the value of is 1. The definition of gene scores (v) states that they anticipate that various parameters will have an impact on its performance α. To reduce the selection pressure of the parameter and make it easier to apply gene scores to various organisms to discover hub genes. The balance of the effect is set to 0.5. The first stage uses edge betweenness centrality to weigh PPI networks with biological documents and topological features. The HITS method is used in the second stage to find the hub gene, which is based on Eq. (6).
Gene Score (w) = α × a (w) + (1 − α) × h (w) (6)
where, α ∈ [0, 1] is employed for modify a ratio of these two scores, where w represents a node. Algorithm to weighted PPI networks [35].
To understand the effectiveness of our method and the significance of identify hub genes, we used some evaluation metric which are described below.
3.1 Functional enrichment analysis
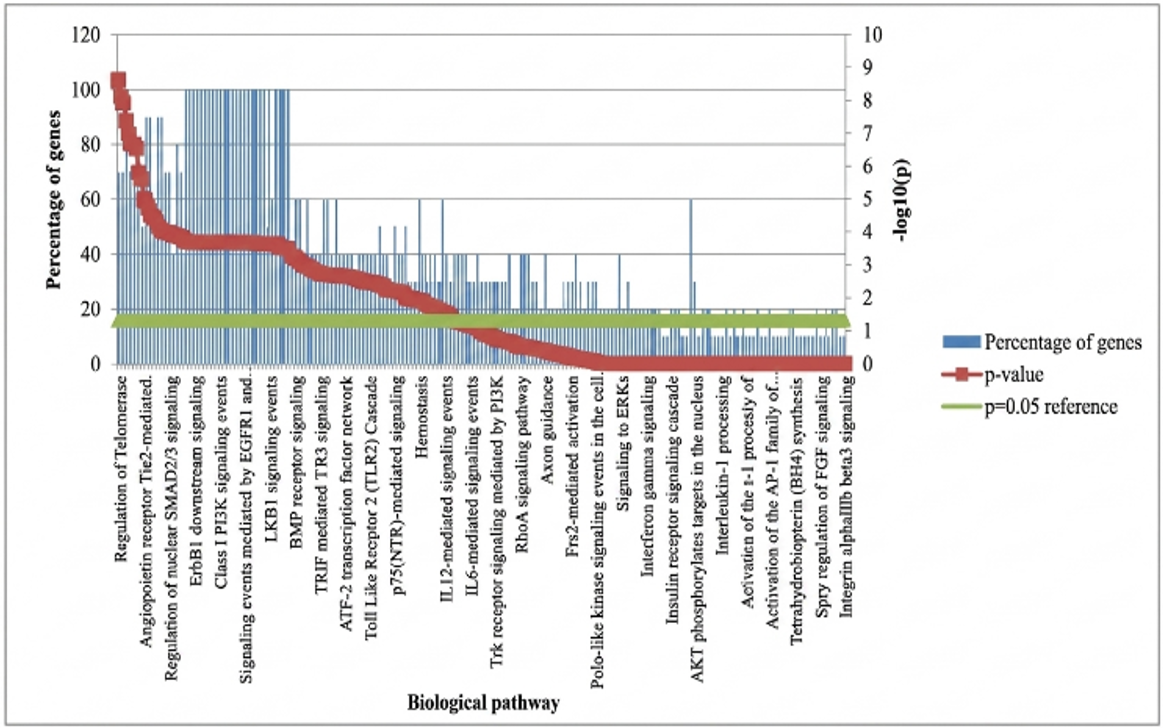
The FunRich analysis is a predominant bioinformatics tool for annotations of genes and their products, in term Biological Pathways (BP) [36]. In general, the tool includes 200 biological pathways, and when the 5hub genes are entered into the tool to be analyzed in the biological pathways the results appeared 34 biological pathways containing 100% of the five hub genes.
3.2 Visualization tool
It is used to visualize biological networks and pathways and integrate them with annotations, gene expression, and other biological data. Although its origins are in biological research, Cytoscape has evolved to analyze sophisticated networks and visualize other networks.
The HITS algorithm's effectiveness needed to be demonstrated, the performance of the algorithm was evaluated by changing the values (α). A thorough sensitivity analysis was performed for (α)values (e.g., 0.2, 0.5, 0.8). The results are now presented, demonstrating the relative stability and robustness of the top-ranked hub genes across these variations. It shows that the hub genes change every time the weight changes for each of h and a (where h called Hub and A called authority) and h shows the biological pathways of the disease and a shows the Topology of the network. The notice that in every change in weight some common genes appear and new genes also appear, this lead to conclusion that it is better to make the value α half to each of each of the h = 0.5 and a = 0.5 to make the network balanced, as shown in Table 2.
Table 2. The weight of both H and A
|
H Ratio (Data GWAS) |
A Ratio |
Top 5 Genes |
|
0.5 |
0.5 |
ITGAM, CSF1R, VCAM1, CCR2, MODY1 |
|
0.6 |
0.4 |
MODY1, ITGAM, CSF1R, VCAM1, CCR2 |
|
0.7 |
0.3 |
CSF1R, VCAM1, CCR2, MODY1, ITGAM |
|
0.8 |
0.2 |
CCR2, MODY1, ITGAM, CSF1R, VCAM1 |
|
0.0 |
1.0 |
VCAM1, CCR2, MODY1, ITGAM, CSF1R |
|
0.9 |
0.1 |
CCR2, MODY1, ITGAM, CSF1R, VCAM1 |
The selection of α = 0.5 was based on stability analysis and ranking consistency. Lower α values overemphasized topological dominance, whereas higher α values excessively prioritized biological annotations. The balanced value (α = 0.5) achieved the highest robustness in preserving recurrent hub genes across multiple iterations and produced the most stable convergence behavior.
Information about protein interactions is crucial. It explains how the biomolecules that genes encode interact with one another. It enables us to anticipate possible treatments and comprehend the intricacies of cellular function. Each node represents a protein, each edge represents an interaction between two proteins, and each edge is weighted by the score. The PPI network and the top five gene scores with the highest connectivity of the genes of interest as the network's hub genes were displayed using the Cytoscape software. That suggests that the HITS algorithm was successful in locating hub genes. Overall, the five hub genes with high connections and most related to disease are CCR2, VCAM1, CSF1R, ITGAM, and MODY1.
A functional enrichment and interaction network study of genes and proteins was conducted using the functional enrichment of Hub Genes). The analysis tab was chosen when conducting the search, and the bar graphs for aspect (biological pathway) were viewed, with the biological pathway being. Several biological pathways were discovered through pathway-based functional enrichment data, suggesting that they are involved in the processes that underlie diabetes. Significant enrichment was seen in pathways pertaining to membrane trafficking and metabolism, lipid modification, lipid reduction, or inhibition. For path analytics, showed a comprehensive profile explanation of the pathogenesis of diabetes at a regular biological level, and identified several biological pathways that play a role in the development of diabetes. It can provide integrated insights to understand the pathogenic mechanism underlying diabetes and its potential new therapeutic targets. The current bioinformatics investigation used the GWAS and ppI Homosapiens data to investigate the overlapping genes and biological pathways for diabetes. Further functional enrichment analysis observed 46 biological pathways which contain all the 5 genes. The significantly enriched pathways included immune response regulation, membrane trafficking, osteoclast differentiation, lipid metabolism, neutrophil degranulation, and oxidative phosphorylation pathways. Statistical significance was evaluated using adjusted p-values and FDR correction. A detailed summary of enriched pathways and corresponding significance values is provided in Supplementary Table 3.
Table 3. CFG classification of the five pivotal genes for vascular aging in diabetic patients
|
Gene |
eQTL |
GWAS |
PPI |
Early_DEG |
Pathology cor (Aβ) |
Pathology cor (Tau) |
CFG |
|
CCR2 |
0 |
0 |
PSEN2, MAPT, APOE |
NA |
- 0.223, ns |
-0.266, ns |
4 |
|
VCAM1 |
1 |
0 |
APP, MAPT, APOE |
NA |
0.871, *** |
0.681, ** |
2 |
|
CSF1R |
5 |
0 |
APP, PSEN1, PSEN2, MAPT, APOE |
NA |
0.110, ns |
0.138, ns |
2 |
|
ITGAM |
1 |
0 |
APP, PSEN2, MAPT |
NA |
0.731, *** |
0.247, ns |
3 |
|
MODY1 |
2 |
0 |
APP, PSEN1, PSEN2, MAPT, APOE |
NA |
-0.170, ns |
-0.485, ns |
3 |
The Validation of Hub Genes related to diabetic vascular aging via Convergent Functional Genomics (CFG). Every piece of evidence has just one CFG point assigned to it (e.g., GWAS indicates that AD genetic variants regulate the expression of the target gene; In diabetic vascular aging, the target gene is expressed differently and interacts physically with CCR2, VCAM1, CSF1R, ITGAM, and MODY1. prior to the development of diabetic vascular aging pathology in mouse models; in Aβ-line diabetic vascular aging mice, the target gene expression is linked to diabetic vascular aging pathology. models and tau line mice with diabetic vascular aging. Between zero and five is the range of CFG points. The CFG ranking places CCR2 as the top gene (four points each), while ITGAM is the second-best gene (three points each), and the third-place genes, with two points apiece, were VCAM1 and CSF1R, while the other two were MODY1. As seen in Table 3.
Software Cytoscape was charity to show the PPI. There were 37219 edges and 5664 nodes in the PPI network, as shown in Figure 5. Overall, the five hub genes that have close relationships were CCR2, VCAM1, CSF1R, ITGAM, and MODY1. Now we all knew that after enough iteration, hub and authority would always converge to a specific value.
Figure 5. An overview of the Protein-Protein Interaction (PPI) with top 5 hub genes
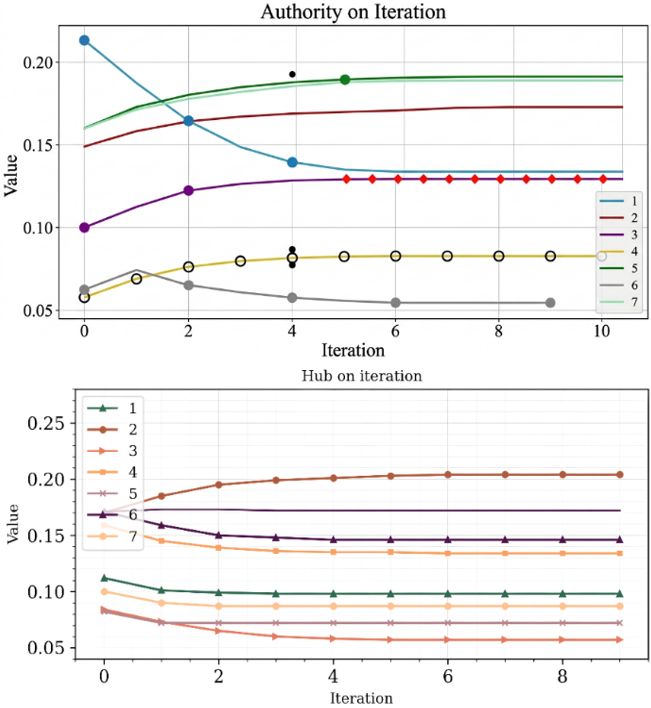
Now we all knew that after enough iteration, hub and authority would always converge to a specific value, as shown in Figure 6.
Figure 6. Performance measurement for convergence
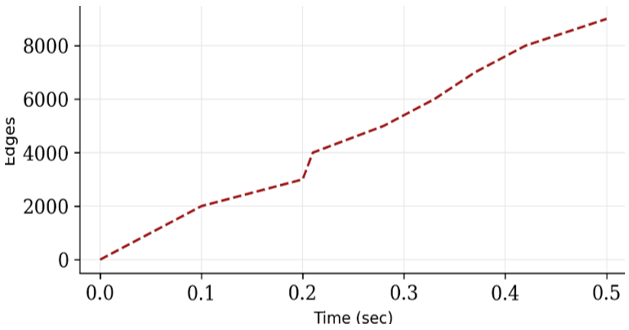
In order to identify the relationship between total edges and computation time, we perform 100 iterations with a varied number of total edges. You can see that the relationship between the inference of edges and computing time is almost linear, which is really good, as shown in Figure 7. Relationship between computation time and total number of edges during HITS algorithm iterations. The x-axis represents the number of network edges, while the y-axis represents computation time in seconds. The results demonstrate an approximately linear relationship between edge size and computational complexity. The edges' connections to one another, which are why it isn't entirely linear, will also, have a small impact on how long it takes to compute. The algorithm demonstrated stable convergence behavior within approximately 500 iterations.
Figure 7. Measure the Hyperlink-Induced Topic Search (HITS) computation
Our study suggests that VCAM1 is a hallmark gene associated with diabetic vascular aging and is linked to disease course, treatment, and prognosis. A comprehensive investigation of the hallmark genes of immune infiltration may shed light on the clinical diagnosis and treatment of the vascular aging process in diabetes.
The strong association between MODY1 and diabetic vascular aging is novel and warrants further investigation.
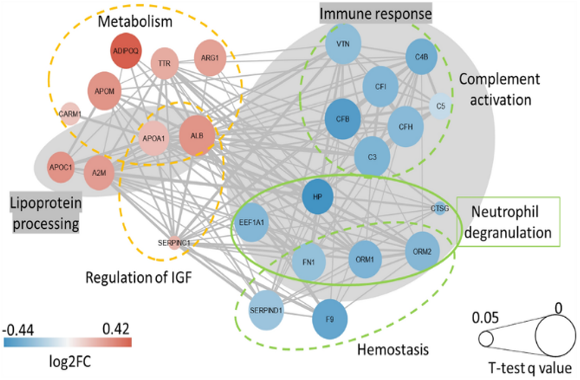
To improve pathway interpretation, enrichment heatmaps and pathway interaction visualizations were generated. These analyses revealed strong associations among immune response, membrane trafficking, and inflammatory signaling pathways, as shown in Figure 8.
Figure 8. Biological pathways of diabetic disease hub genes
In conclusion, our study provides more insight into how VSMC senescence contributes to diabetes's development of vascular aging. We discovered five important genes that are strongly linked to it, using bioinformatics methods to study vascular aging in diabetes. Understanding the basic processes that underline cellular life is significant for identifying key genes. This article introduced a novel computer method to determine important proteins via a weighted PPI network and the HITS algorithms. A weighted PPI network is used to discover essential proteins using biomedical information and network topology. DEGs with upregulation showed a strong link with osteoclast differentiation, lysosome, neutrophil degranulation, and immune response. Although the proposed computational framework demonstrated robust performance, experimental validation using in vitro cellular models and in vivo diabetic vascular aging models remain necessary to confirm the biological significance of the identified hub genes.
In contrast, DEGs showed a major connection to ribosome, translation, peptide production, and oxidative phosphorylation through downregulation. Consequently, we discovered CCR2, VCAM1, CSF1R, ITGAM, and MODY1 as essential genes in diabetes determined by PPI network analysis. This HITS method is used in this study for extensive bioinformatics investigation to deepen our accepting of the molecular basis from diabetes. The identified hub genes may provide candidate biomarkers for future experimental validation and therapeutic investigations in diabetic vascular aging.
Our study provides a computational strategy for identifying hub genes in diabetic vascular aging. We discovered five key genes (CCR2, VCAM1, CSF1R, ITGAM, MODY1) using a weighted HITS algorithm that integrates PPI network topology with gene expression and functional annotations. Functional analysis linked these genes to critical pathways like immune response and membrane trafficking. Limitations this study has certain limitations. The analysis relies on pre-existing datasets, and the findings require validation through in vitro or in vivo experiments. The PPI network, while comprehensive, may contain false positives and lack temporal/spatial dynamic information. Despite the promising findings, this study has several limitations.
The analysis relied primarily on publicly available datasets and computational predictions without experimental validation. In addition, the PPI network may contain incomplete or false-positive interactions that could influence gene ranking performance.
Future work will focus on experimental validation of the identified hub genes. We also plan to incorporate additional data layers, such as epigenomics and single-cell RNA sequencing data, and explore more complex graph neural network models to capture the dynamic nature of biological interactions.
[1] Adu, R.O., Boateng, K.K., Asiedu, W. (2024). HITS vs. pagerank: A comparative analysis of web search algorithms. International Journal of Computer Applications, 186(51): 32-36. https://doi.org/10.5120/ijca2024924177
[2] Lee, K.Y., Leung, K.S., Ma, S.L., So, H.C., Huang, D., Tang, N.L.S., Wong, M.H. (2020). Genome-wide search for SNP interactions in GWAS data: Algorithm, feasibility, replication using schizophrenia datasets. Frontiers in Genetics, 11: 1003. https://doi.org/10.3389/fgene.2020.01003
[3] Zhang, Z.R., He, X.L., Sun, Y.X., Li, J.T., Sun, J. (2025). Type 2 diabetes mellitus: A metabolic model of accelerated aging-multi-organ mechanisms and intervention approaches. Aging and Disease, 17(3): 1399-1422. https://doi.org/10.14336/AD.2025.0233
[4] Chen, S., Lian, J., Mathew, R., Lu, M., Chen, T., Sarwar, A.I., Levitan, I., Lee, M., Chen, J.W. (2024). Vascular aging: Implications, mechanisms, and interventions. Aging Research, 2(4): 9340039. https://doi.org/10.26599/AGR.2025.9340039
[5] Wang, S., Wang, X., Chen, J., Wang, M., Zhang, C. (2024). Identification of key genes and biological pathways associated with vascular aging in diabetes based on bioinformatics and machine learning. Aging, 16(11): 9369-9385. https://doi.org/10.18632/aging.205870
[6] Li, Q.X., Li, P.S., Xu, Z.G., Lu, Z.Y., Yang, C., Ning, J. (2024). Association of diabetes with cardiovascular calcification and all-cause mortality in end-stage renal disease in the early stages of hemodialysis: A retrospective cohort study. Cardiovascular Diabetology, 23(1): 259. https://doi.org/10.1186/s12933-024-02318-8
[7] Li, Y.W., Liu, Y.F., Liu, S.W., Gao, M.Q., Wang, W.T., Chen, K.J., Huang, L.Q., Liu, Y. (2023). Diabetic vascular diseases: Molecular mechanisms and therapeutic strategies. Signal Transduction and Targeted Therapy, 8(1): 152. https://doi.org/10.1038/s41392-023-01400-z
[8] Zakir, M., Ahuja, N., Surksha, M.A., Sachdev, R., et al. (2023). Cardiovascular complications of diabetes: From microvascular to macrovascular pathways. Cureus, 15(9): e45835. https://doi.org/10.7759/cureus.45835
[9] Gupta, S., Hernandez, G., Raman, P. (2025). Cellular and molecular mechanisms of VSMC phenotypic switching in type 2 diabetes. Cells, 14(17): 1365. https://doi.org/10.3390/cells14171365
[10] Chen, H.X., Peng, C.X., Fang, F., Li, Y.H., Liu, X.R., Hu, Y., Wang, G.X., Liu, X.H., Shen, Y. (2025). Angiogenesis within atherosclerotic plaques: Mechanical regulation, molecular mechanism and clinical diagnosis. Mechanobiology in Medicine, 3(1): 100114. https://doi.org/10.1016/j.mbm.2025.100114
[11] Zhao, X.Y., Yang, X.Y., Lin, Y.M., Lei, R.L., et al. (2025). Mechanisms of aging in the cardiovascular system: Challenges and opportunities. Frontiers in Immunology, 16: 1635736. https://doi.org/10.3389/fimmu.2025.1635736
[12] Ranganathan, Y., Nazar, S., Krishnan, R.S., Dinakarkumar, Y., Varadarajan, V., Sebastian, L., Rethinam, B. (2024). Understanding integrative approach of translational bioinformatics on cardiovascular disease: Myocardial ischemia. Egyptian Journal of Medical Human Genetics, 25(1): 153. https://doi.org/10.1186/s43042-024-00622-2
[13] Yuan, R.Q., Zhang, J., Zhou, J., Cong, Q. (2025). Recent progress and future challenges in structure-based protein-protein interaction prediction. Molecular Therapy, 33(5): 2252-2268. https://doi.org/10.1016/j.ymthe.2025.04.003
[14] Acharya, D., Dutta, T.K. (2021). Elucidating the network features and evolutionary attributes of intra- and interspecific protein–protein interactions between human and pathogenic bacteria. Scientific Reports, 11(1): 190. https://doi.org/10.1038/s41598-020-80549-x
[15] Bai, L., Su, J.C. (2026). Artificial Intelligence Virtual Organoids (AIVOs). Bioactive Materials, 59: 45-68. https://doi.org/10.1016/j.bioactmat.2025.12.030
[16] Mohammed, H.A.A., Kasim Jizany, A.A., Mahmood, I.M., Kadhim, Q.K. (2024). Predicting Alzheimer’s disease using a modified grey wolf optimizer and support vector machine. Ingenierie Des Systemes d’Information, 29(2): 669-676. https://doi.org/10.18280/isi.290228
[17] Sang, Y.T., Xu, L.J., Bao, Z.H. (2024). Development of artificial transcription factors and their applications in cell reprograming, genetic screen, and disease treatment. Molecular Therapy, 32(12): 4208-4234. https://doi.org/10.1016/j.ymthe.2024.10.029
[18] Soleymani, F., Paquet, E., Viktor, H., Michalowski, W., Spinello, D. (2022). Protein–protein interaction prediction with deep learning: A comprehensive review. Computational and Structural Biotechnology Journal, 20: 5316-5341. https://doi.org/10.1016/j.csbj.2022.08.070
[19] Xu, W.Z., Neal, E.S., Barlow, N., Thompson, P., Borges, K. (2025). Altered glut4, IRAP, and brain insulin signaling in a mouse model of epilepsy and contributions to glucose transport in neurons and astrocytes. Journal of Neurochemistry, 169(12): e70320. https://doi.org/10.1111/jnc.70320
[20] Merumba, S.B., Ahmed, H.O., Fu, D., Yang, P.F. (2025). Recent advances and application of machine learning for protein–protein interaction prediction in rice: Challenges and future perspectives. Proteomes, 13(4): 54. https://doi.org/10.3390/proteomes13040054
[21] Kouba, P., Kohout, P., Haddadi, F., Bushuiev, A., Samusevich, R., Sedlar, J., Damborsky, J., Pluskal, T., Sivic, J., Mazurenko, S. (2023). Machine learning-guided protein engineering. ACS Catalysis, 13(21): 13863-13895. https://doi.org/10.1021/acscatal.3c02743
[22] Hein, Z.M., Guruparan, D., Okunsai, B., Che Mohd Nassir, C.M.N., Ramli, M.D.C., Kumar, S. (2025). AI and machine learning in biology: From genes to proteins. Biology, 14(10): 1453. https://doi.org/10.3390/biology14101453
[23] Abdulbaqi, H., Taha, A.M.S., Mohammed, A.R., Kanaan, H.Z.Q., Kadhim, Q.K. (2024). Detection and localization of wrist fractures in x-ray imagery using deep learning teaching. Review of Computer Engineering Research, 11(3): 85-98. https://doi.org/10.18488/76.v11i3.3850
[24] Khan, M., Mello, G.B.M., Habib, L., Engelstad, P., Yazidi, A. (2024). HITS-based propagation paradigm for graph neural networks. ACM Transactions on Knowledge Discovery from Data, 18(4): 1-23. https://doi.org/10.1145/3638779
[25] Zhao, Q.J., Jiao, X. (2022). Orientation algorithm for PPI networks based on network propagation approach. Journal of Biosciences, 47(3): 44. https://doi.org/10.1007/s12038-022-00284-5
[26] Yin, H.C., Duo, H.R., Li, S., Qin, D., et al.(2025). Unlocking biological insights from differentially expressed genes: Concepts, methods, and future perspectives. Journal of Advanced Research, 76: 135-157. https://doi.org/10.1016/j.jare.2024.12.004
[27] Zhang, Z.H., Luo, Y.C., Hu, S., Li, X.Y., Wang, L., Zhao, B.H. (2020). A novel method to predict essential proteins based on tensor and HITS algorithm. Human Genomics, 14(1): 14. https://doi.org/10.1186/s40246-020-00263-7
[28] Jha, K., Saha, S., Singh, H. (2022). Prediction of protein–protein interaction using graph neural networks. Scientific Reports, 12(1): 8360. https://doi.org/10.1038/s41598-022-12201-9
[29] Olushola, A., Adesiyan, A., Ojo, T., Alao, V. (2024). Bioinformatics and computational biology: Exploring the role of biomathematics in bioinformatics, including the analysis of biological data and the development of computational tools. Preprints, 1-60. https://doi.org/10.20944/preprints202411.0570.v1
[30] Chen, J.B., Chang, C.H. (2025). Using hyperlink-induced topic search algorithm to optimize content placement in multimedia content delivery network. Journal of Circuits, Systems and Computers, 34(05): 2550130. https://doi.org/10.1142/S0218126625501300
[31] Jaiganesh, S., Aravind Babu, L.R. (2024). Enhancing link evaluation through a coordinated structure: A comparative analysis of pagerank and HITS Algorithms. Nanotechnology Perceptions, 20(S7). https://doi.org/10.62441/nano-ntp.v20iS7.3
[32] Alwan, O.F., Kadhim, Q.K., Issa, R.B., Ahmed, S.T. (2023). Early detection and segmentation of ovarian tumor using convolutional neural network with ultrasound imaging. Revue d’Intelligence Artificielle, 37(6): 1503-1509. https://doi.org/10.18280/ria.370614
[33] Cai, F.H., Qian, F.C., Li, B.L., Li, L.D., et al. (2025). DiabetesOmic: A comprehensive multi-omics diabetes database. Computational and Structural Biotechnology Journal, 27: 2147-2154. https://doi.org/10.1016/j.csbj.2025.05.008
[34] Nieman, D.C., Sakaguchi, C.A., Pelleigrini, M., Thompson, M.J., Sumner, S., Zhang, Q.B. (2023). Healthy lifestyle linked to innate immunity and lipoprotein metabolism: A cross-sectional comparison using untargeted proteomics. Scientific Reports, 13(1): 16728. https://doi.org/10.1038/s41598-023-44068-9
[35] Tsare, E.P.G., Klapa, M.I., Moschonas, N.K. (2024). Protein–protein interaction network-based integration of GWAS and functional data for blood pressure regulation analysis. Human Genomics, 18(1): 15. https://doi.org/10.1186/s40246-023-00565-6
[36] Hou, J.Y., Li, Y.B., Zhang, Y., Yang, N., Chen, B., Ma, G.Y., Zhu, N.Q. (2025). Integrated network pharmacology reveals the mechanism of action of Xianlinggubao prescription for inflammation in osteoarthritis. BMC Complementary Medicine and Therapies, 25: 190. https://doi.org/10.1186/s12906-025-04928-5