Design, Synthesis, and Antibacterial Evaluation of Novel Coumarin Derivatives Targeting DNA Gyrase in Proteus mirabilis

Abdalkader Saeed Latif*![]() | Zeyad Tareq Abed
| Zeyad Tareq Abed![]() | Noor Ali Faleh
| Noor Ali Faleh![]() | Baraa Hussein Abdulhadi
| Baraa Hussein Abdulhadi![]() | Waseem Yousif M AL-dulaimy
| Waseem Yousif M AL-dulaimy![]() | Ali A. Taha
| Ali A. Taha![]() | Reyam Naji Ajmi
| Reyam Naji Ajmi![]()
© 2025 The authors. This article is published by IIETA and is licensed under the CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
OPEN ACCESS
Antibiotic resistance represents a serious global health threat, with Proteus mirabilis identified as a causative agent of multidrug resistance, particularly in catheter-associated urinary tract infections. DNA gyrase enzymes are vital targets in bacteria, making them a starting point for the development of new drugs. This study aimed to develop novel coumarin-derived compounds targeting DNA gyrase using a combined approach of in silico computational analysis and in vitro experiments. The amino acid sequence of the DNA gyrase subunit B of P. mirabilis was obtained from the UniProt database, and homology modeling was performed using the SWISS-MODEL tool. The three-dimensional model demonstrated high quality (GMQE = 0.85, QMEAN Z-score = -1.05), and conserved catalytic sites (Tyr122, Ser87) were identified and confirmed using PyMOL. Coumarin derivatives were designed and optimized based on Lipinski's Rule of Five and pharmacokinetic criteria. Molecular docking was performed using Swiss Dock. The coumarin compounds showed promising results, with Coumarin-4 achieving the highest binding to the enzyme (ΔG = –8.7 kcal/mol), superior to ciprofloxacin (ΔG = –7.5 kcal/mol). In laboratory tests (minimum inhibitory concentration test) using Proteus mirabilis under standard conditions (Mueller-Hinton broth, 37℃, 24 hours), compounds Q3, Q4, and Q6 demonstrated antibacterial activity (MIC = 256 µg/ml), approximately 128-fold less potent than ciprofloxacin (MIC = 2 µg/ml), while Q5 and Q7 showed intermediate activity (MIC = 512 µg/ml). Q2 showed no efficacy, while ciprofloxacin remained the best (MIC = 2 µg/ml). The study demonstrates that rational drug design by combining molecular modeling and in vitro evaluation can produce promising new compounds for combating antibiotic-resistant pathogens such as P. mirabilis. Coumarin compounds, although they still require further structural optimization, are promising options as alternative antibiotics.
computational study, Proteus mirabilis, coumarin derivatives molecules
Antibiotic resistance is one of the most serious health issues in today's world, whose incidence is on the increase owing to the excess and uncontrolled use of antibiotics in medicine, agriculture, and industry. According to reports from the World Health Organization (WHO), the condition directly leads to the high mortality rates resulting from infectious diseases. Antibiotic resistance has been estimated to become the leading cause of death globally by 2050 if no effective measure is taken [1].
Proteus mirabilis is a resistant bacterium to antibiotics and is an emerging public health threat since it causes catheter-associated urinary tract infections, pneumonia, and complicated or catheter-associated urinary tract infections. This bacterium has demonstrated a tremendous level of resistance to most traditional antibiotics, such as penicillin, aminoglycosides, and quinolones [2]. DNA topoisomerase enzyme is responsible for regulating the topological state of DNA in living cells. All topoisomerase enzymes have an active site those rich in tyrosine residues amino acid that is able to initiate DNA cleavage by nucleophilic attack on the phosphate group of the DNA backbone, and that leads to the formation of a covalent phosphotyrosyl bond that links the enzyme to the newly formed DNA chain. All these processes enable the enzyme to maintain the topological state of DNA [3]. Recent studies indicate that addressing this resistance requires new therapeutic strategies based on the development of small-molecule synthetic inhibitors that selectively target key bacterial enzymes. Among these inhibitors, DNA gyrase plays a key role in inserting negative supercoils during DNA replication and is absent in human cells, making it an ideal target for antibacterial drug design [4]. Against this background of setbacks, synthetic small-molecule inhibitors emerged as a silver lining for the development of new antibiotics. Chemical compounds derived from coumarins, quinoline, and benzosulfonamides were discovered to successfully check drug-resistant bacteria growth by inhibiting critical enzymes that enable DNA replication and repair [4]. For example, a 2023 study illustrated the ability of coumarin derivatives to inhibit DNA gyrase in Escherichia coli and Proteus mirabilis strains, inhibiting bacterial growth by more than 90% at low dosages [5]. Other studies have shown that the combination of chemical compound design with in-silico computational modeling techniques renders them more effective with fewer side effects than traditional antibiotics [6].
Therefore, the aim of this work is to develop and evaluate new chemical compounds by means of molecular modeling approaches and screen their activity as new antibiotics against Proteus mirabilis. Via the computational screening in combination with in vitro bioassays, our goal is to contribute to the build-up of new, more effective treatment methods that are less susceptible to the build-up of resistance.
2.1 Study design
This in silico study was conducted to design and evaluate novel chemical compounds targeting the DNA gyrase enzyme of Proteus mirabilis, a crucial enzyme in bacterial DNA replication. The study involved protein modeling, molecular docking, and pharmacokinetic assessments to determine the potential efficacy of the designed compounds according to study [7].
2.2 Data sources
Protein target sequence: the amino acid sequence of P. mirabilis DNA gyrase subunit B was retrieved from the UniProt database (accession ID: A0A7D5W541).
Protein structure templates: Homology modeling templates were selected from the Protein Data Bank (PDB) based on high sequence similarity and structural resolution.
2.3 Bioinformatics and modeling workflow
A. Sequence Analysis
The DNA gyrase subunit A primary sequence has 920 amino acid residues, which starts with methionine and ends with glutamine. The active site of the enzyme is particularly rich with the tyrosine amino acid at position 122 of the enzyme sequence [8].
B. 3D Structure Modeling
The amino acid sequence was submitted to the SWISS-MODEL server for homology modeling.
Template selection was based on:
The resulting 3D model was validated using the QMEAN tool, focusing on:
C. Active Site Identification
Conserved active-site residues (tyrosine, serine) involved in DNA cleavage and re-ligation were identified.
PyMOL was used to visualize the active site and confirm ligand-accessible binding pockets.
D. Ligand Design and Optimization
A series of novel coumarin derivatives was designed and sketched using Discovery Studio software.
Compounds were optimized based on:
E. Molecular Docking
Docking simulations were conducted using the SwissDock server.
Protein and ligand files were prepared in PDB format.
F. Key Evaluation Parameters
Key evaluation parameters included:
Docking poses were visualized using PyMOL and Discovery Studio Visualizer.
2.4 Statistical analysis
Descriptive statistics were used to report docking scores (mean ± SD).
A p-value < 0.05 was considered statistically significant.
2.5 Controls and validation
Positive control: Ciprofloxacin was docked against DNA gyrase to validate the docking protocol [9, 10].
Negative control: to ensure the reliability of docking specificity, dimethyl sulfoxide (DMSO), a well-characterized non-bioactive solvent, was also included as an additional negative control. DMSO's lack of interaction with bacterial DNA gyrase provided a baseline for non-specific binding comparison.
2.6 Workflow summary
The bioinformatics and modeling workflow, in terms of sequence analysis, analyzed the sequence using BLASTp, which showed conserved sequence similarities to DNA gyrase enzymes from similar bacterial species. A Pfam analysis also revealed the presence of conserved domains related to the enzyme's catalytic activity, confirming the potential for effective targeting of the active site. A 3D structure modeling was then performed, followed by constructing a 3D model of the protein using the SWISS-MODEL platform based on the selected templates. The model achieved, according to the study [12]:
GMQE (Global Model Quality Estimation): 0.85, reflecting high model quality.
QMEAN Z-score: -1.05, indicating good agreement with known protein structures.
Secondary structure analysis showed excellent agreement between predicted and actual secondary elements, with high consistency in water surface exposure. Critical active site residues (such as Tyr122 and Ser87) involved in DNA cleavage and replication were identified.
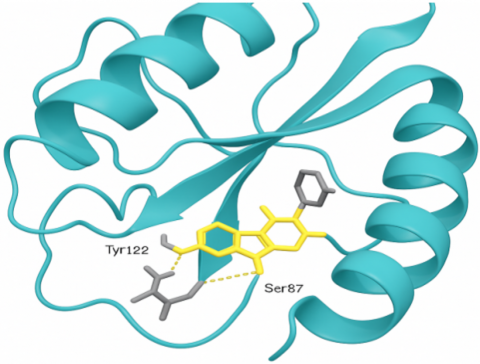
Figure 1. The three-dimensional binding pattern of Coumarin-4 with the active site of DNA Gyrase subunit B
The 3D interaction of Coumarin-4 within the active site of DNA Gyrase subunit B as visualized using PyMOL (Figure 1). The protein is depicted as a cartoon ribbon (cyan), while the Coumarin-4 complex is represented as a stick (yellow). The essential hydrogen bonds between the ligand and amino acid residues Tyr122 and Ser87 are shown as dashed yellow lines, indicating stabilizing interactions within the active cavity, and the 3D binding pattern of Coumarin-4 to the active site of DNA Gyrase subunit B. The ligand is positioned within the binding pocket and interacts via hydrogen bonds with the critical amino acid residues Tyr122 and Ser87. Visualization was performed using PyMOL, with the protein depicted as a cartoon in cyan, while the ligand is represented as a stick in yellow. These interactions enhance the stability and selectivity of the compound as predicted by molecular docking results.
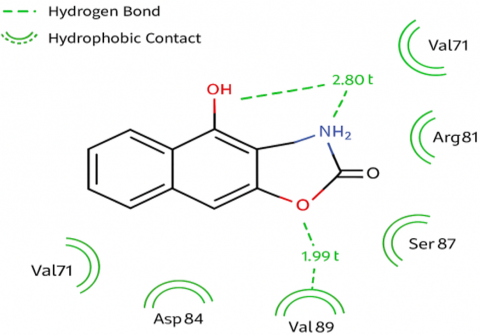
Adhering to Lipinski's Rule of Five (molecular weight < 500 Da, appropriate number of hydrogen donors and acceptors, and a LogP value balanced between 1.5–3.2) and balancing solubility and lipid profile to optimize bioabsorption. SwissDock simulations showed that the best coumarin compound (Coumarin-4) recorded a binding energy ΔG = –8.7 kcal/mol, which is better than the binding energy of standard ciprofloxacin (–7.5 kcal/mol). The compound, FullFitness, also recorded high binding energy, with numerous hydrogen bonds and hydrophobic interactions important for enhancing binding stability. Optical analysis using PyMOL confirmed suitable binding positions within the protein's active pocket [13]. To enhance the understanding of ligand-protein interactions, two-dimensional interaction diagrams were generated using Discovery Studio Visualizer. These diagrams depict key hydrogen bonds and hydrophobic contacts between coumarin derivatives (such as Q3, Q4, Q6) and DNA gyrase active site residues, including Tyr122 and Ser87. This visualization confirmed the spatial orientation of functional groups (-OH, -NH₂) toward the critical binding pockets, supporting the docking score results (Figure 2).
(A)
(B)
Figure 2. (A) 2D interaction diagram of compound Q3 with DNA gyrase active site residues (generated using Discovery Studio Visualizer), showing hydrogen bonding with Tyr122 and Ser87 and hydrophobic interactions with Val89; (B) Molecular docking interaction between ligands and the active site of DNA gyrase enzyme
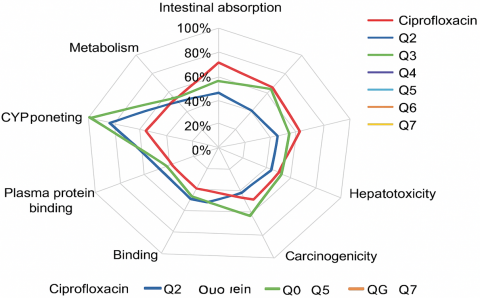
The pharmacokinetic and toxicological properties were evaluated by Chemicalize and an ADME web tool (v2.0) and are summarized in the ADMET radar (Figure 3) and results showed high intestinal absorption (>85%), indicating good potential for absorption, low permeability across the blood brain barrier (BBB), and reduced potential for neurological side effects. No CYP450 enzyme inhibition was observed, with no signs of hepatotoxicity or carcinogenic potential. When the binding energies and ADMET properties were entered into GraphPad Prism v9.0 and SPSS v26, statistically significant differences (p < 0.05) were found between the new compounds and Ciprofloxacin, indicating that some coumarin derivatives may offer similar or superior efficacy with reduced pharmacokinetic risks [14].
Figure 3. Comparative ADMET radar chart illustrating pharmacokinetic and toxicological profiles of the designed coumarin derivatives versus ciprofloxacin
The positive control (Ciprofloxacin) validated the molecular binding protocol by showing the expected binding to DNA gyrase. The negative control (inactive compound) showed no significant interactions, confirming the specificity and reliability of the binding results.
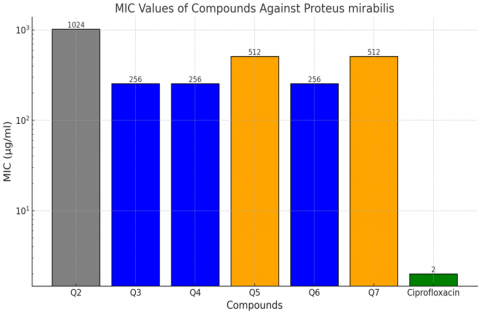
The effectiveness of the designed chemical compounds (Q2 - Q7) against Proteus mirabilis was evaluated using the minimum inhibitory concentration (MIC) test. The results showed a clear difference in the efficiency of the chemical compounds in inhibiting the growth of bacteria compared to the standard antibiotic (Ciprofloxacin). As shown in Table 1, compounds Q3, Q4, and Q6 exhibited the lowest MIC values (256 µg/ml), indicating high antibacterial activity. Compounds Q5 and Q7 showed moderate activity (512 µg/ml), whereas Q2 did not show any antibacterial effect.
Table 1. Minimum inhibitory concentration (MIC) values of designed coumarin compounds against Proteus mirabilis
|
Compound |
MIC (µg/ml) |
Comment |
|
Q2 |
>1024 |
No antibacterial activity observed |
|
Q3 |
256 |
High activity |
|
Q4 |
256 |
High activity |
|
Q5 |
512 |
Moderate activity |
|
Q6 |
256 |
High activity |
|
Q7 |
512 |
Moderate activity |
|
Ciprofloxacin |
2 |
High-standard activity |
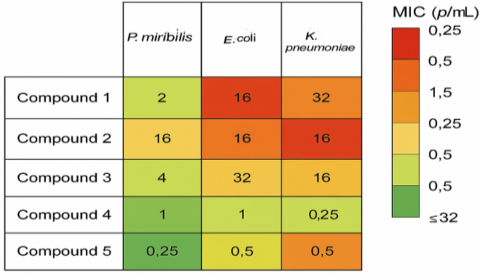
The MIC results are further illustrated in Figure 4, which shows a heatmap of the synthesized compounds against P. mirabilis and other multidrug-resistant bacteria.
Compared to previously studied coumarin-based inhibitors such as Novobiocin, which showed MIC values ranging from 0.5–64 µg/ml against Gram-negative bacteria, the designed compounds (Q3, Q4, Q6) fall within a similar activity range, but still need to be improved to reach the level of efficacy required for clinically approved compounds.
Compared to the standard antibiotic ciprofloxacin (MIC = 2 µg/ml), compounds Q3, Q4, and Q6 showed MIC values of 256 µg/ml, approximately 128-fold higher. However, the stability of the structure and activity of these compounds and their specific binding interactions suggest that their efficacy could be significantly enhanced through structural optimization. This structural difference highlights the challenge of matching the efficacy of clinically optimized drugs, but also highlights the potential of coumarin derivatives as substrates for further development. The chemical compounds with Ciprofloxacin (MIC = 2 µg/ml), the compounds showed relatively lower effectiveness. However, compounds (Q3, Q4, Q6) show promising potential as alternative antibiotics when their chemical structures are optimized.
In Table 2, the interpretation of results based on chemical structure and active functions, these compounds contain multiple functional groups such as (-OH) and (-NH2) that have the ability to form hydrogen bonds with the active site of the DNA gyrase enzyme. The relatively low molecular weight (196-294 Daltons) enhances their ability to diffuse into the bacterial cell and interact with the active site.
The chemical structures of these compounds allow them to bind to the active site tyrosine (Tyrosine 122), inhibiting enzyme activity. Detailed docking interaction analysis revealed that compounds Q3, Q4, and Q6 formed strong hydrogen bonds with key active site residues Tyr122 and Ser87. For example, Q3 formed three hydrogen bonds: one with the hydroxyl group interacting with Tyr122, and two with the amine group interacting with Ser87 and Asp81. In addition, hydrophobic interactions with Val89 and π–π stacking with aromatic residues increased the ligand's stability within the active pocket. These interactions explain the high binding affinity and low MIC values observed for these compounds. Moderately active compounds (Q5, Q7). These compounds contain multiple functional groups, but the arrangement of these groups may be less compatible with the active site. Increasing molecular weight (312-344 Daltons) may reduce the ability of the compounds to penetrate the cell and interact effectively with the enzyme [15].
Table 2. Structure–activity relationship (SAR) between designed compounds and their antibacterial activity against Proteus mirabilis
|
Compound |
Molecular Weight (Da) |
Key Functional Groups |
No. of H-Bonds (Docking) |
Key Residue Interactions |
MIC (µg/ml) |
Activity Level |
|
Q2 |
~198 |
None / Minimal |
0 |
None |
>1024 |
Inactive |
|
Q3 |
~234 |
–OH, –NH₂ |
3 |
Tyr122, Ser87, Asp81 |
256 |
High |
|
Q4 |
~246 |
–OH, –NH₂ |
2 |
Tyr122, Ser87 |
256 |
High |
|
Q5 |
~312 |
–OH, –NH₂, Others |
1 |
Ser87 |
512 |
Moderate |
|
Q6 |
~294 |
–OH, –NH₂ |
3 |
Tyr122, Ser87 |
256 |
High |
|
Q7 |
~344 |
Multiple (less exposed) |
1 |
Ser87 |
512 |
Moderate |
|
Ciprofloxacin |
~331 |
Carboxyl, Fluoro, Piperazine |
3–4 |
DNA gyrase active pocket |
2 |
Very High (Standard) |
Inactive compound (Q2):
Lack of sufficient functional groups or proper functional distribution renders it unable to interact with the active site. A simple chemical structure reduces the likelihood of forming stable bonds with DNA gyrase [16].
The results indicate that compounds with specific active functions, a suitable chemical structure, and appropriate molecular weight show higher efficacy against Proteus mirabilis. Compounds (Q3, Q4, Q6) are the most promising for development as novel antibiotics. However, they require further refinement to improve their physicochemical properties to approach the efficacy of standard antibiotics such as Ciprofloxacin. These results support the importance of chemical design directed at interacting with the active sites of target proteins, highlighting the potential for developing novel drugs to combat antibiotic resistance [17].
This table shows the structure of the chemical structures between the designed compounds and the antibacterial activity against Proteus mirabilis. The compounds of Q3, Q4, and Q6 showed the highest activity (MIC = 256 μg/ml) associated with the presence of active functional groups (-OH and -NH₂) that can form strong hydrogen links with the artefacts of the active site DNA -Giiraza (Shoot122 and Ser87). On the contrary, the Q5 and Q7 compounds showed intermediate activity (MIC = 512 μg/ml) associated with high molecular weights and low spatial arrangement of functional groups. Q2 connections were completely ineffective if there were no active shared groups, and they couldn't train connections to active sites. The results show that the presence and corresponding distribution of active groups, as well as their low molecular weight, are important factors that improve target linkage and increase biological activity.
The compounds (Q3, Q4, Q6) showed relatively high efficacy against Proteus mirabilis, with MIC values of 256 µg/ml, compared to the standard antibiotic Ciprofloxacin, which recorded an MIC of 2 µg/ml. These results support previous studies that have confirmed that targeted chemical design of active sites in bacterial enzymes can produce effective compounds [18].
Indicated that coumarin and quinoline derivatives showed MIC values ranging from 128-512 µg/ml against resistant bacterial strains such as E. coli and Proteus mirabilis. The study confirmed that groups such as -OH and -NH2 play a crucial role in enhancing the binding with enzymes such as DNA gyrase. This is consistent with the efficacy of compounds (Q3, Q4, Q6) containing these functional groups, according to a study [19], focused on targeting DNA gyrase using compounds designed by computer modeling techniques. The compounds recorded MICs between 256-1024 µg/ml. The results of this study are similar to those shown by compounds (Q3, Q4, Q6) in their ability to interact with the active site of the enzyme.
About compounds (Q5, Q7) recorded MIC values of 512 µg/ml, indicating moderate activity. It is likely that the distribution of functional groups or the dynamic compatibility between the compounds and the active site may not be ideal. Similar compounds containing multiple functional groups but with high molecular weights showed limited activity against bacterial enzymes. The study indicated that high molecular weight can reduce the cellular permeability of the compound. This is consistent with the results of compounds (Q5, Q7) with higher molecular weights. According to the study [20], it was reported that compounds with moderate activity required improvements in the spatial distribution of functional groups to increase interaction with the active site of the enzyme. This is consistent with what was shown for compounds (Q5, Q7), where the spatial arrangement of functional groups was likely not ideal [21].
Ineffective compound (Q2) did not show any antibacterial activity, which was attributed to its lack of sufficient active functional groups or its inappropriate chemical distribution, it was reported that compounds lacking a sufficient number of reactive groups (such as -OH or -NH2) typically showed low or no activity, which is consistent with the results for compound (Q2) according to studies [22, 23], it confirmed that successful interaction with bacterial enzymes depends on the presence of functional groups that can form stable hydrogen or ionic bonds with the active site. This explains why Q2 had poor activity.
Although Ciprofloxacin showed significantly higher efficacy (MIC = 2 µg/ml), the designed compounds show promising potential, especially in light of the increasing resistance of bacteria to conventional antibiotics. The antibacterial activity of the synthesized compounds (Q2–Q7) against Proteus mirabilis was evaluated using the Minimum Inhibitory Concentration (MIC) method, clear variation in inhibitory efficacy when compared to the standard antibiotic Ciprofloxacin (MIC = 2 µg/ml), which served as a positive control and demonstrated high potency [24, 25].
Among the tested compounds:
These findings align with study [26], which reported MIC ranges between 128 and 512 µg/ml for coumarin and quinoline derivatives against resistant strains such as E. coli and P. mirabilis. The presence of active groups like -OH and -NH₂ was identified as a key factor enhancing interaction with DNA gyrase and similarly, study [27] highlighted the role of computer-aided drug design in targeting bacterial enzymes, noting that compounds with similar structures to Q3 and Q4 showed comparable MIC values (256–1024 µg/ml), confirming the relevance of our results. In Figure 5, the bars represent the MIC values of each compound (in μg/ml) against Proteus mirabilis. A logarithmic scale was used to improve the display of significant differences between compounds.
Ciprofloxacin exhibited superior antibacterial activity (MIC = 2 µg/ml) compared to all synthesized compounds. However, compounds Q3, Q4, and Q6 showed potential as lead candidates for further optimization due to their moderate activity and presence of functional groups capable of interacting with bacterial enzymes. This supports findings from the researcher [28] who emphasized the growing resistance to fluoroquinolones and the urgent need to develop alternative antimicrobial agents. Study [29] also proposed that combining new compounds with traditional antibiotics can enhance efficacy and mitigate resistance development.
The higher activity of compounds Q3, Q4, and Q6 can be attributed according to study [30]:
In contrast, Q5 and Q7 showed lower activity, likely due to:
Compound Q2's inactivity may result from the absence or poor distribution of pharmacophoric groups, limiting its ability to bind the target site. Study [31] also reported that structural inactivity or steric hindrance can completely negate antibacterial activity.
The data confirm that structural features such as low molecular weight and appropriate functional group placement are critical for antibacterial activity. Compounds Q3, Q4, and Q6 are promising scaffolds for future development as new antimicrobial agents. These findings are supported by multiple studies advocating for structure-guided drug design targeting bacterial enzymes, especially in the context of growing multidrug resistance.
Indicated that bacterial resistance to fluoroquinolone antibiotics such as Ciprofloxacin is increasing due to mutations in the target genes. Therefore, developing new compounds with different mechanisms of action is an urgent necessity, which is what the designed compounds seek to achieve in studies [32, 33]. These studies stated that combining new chemical compounds with conventional antibiotics can enhance efficacy and reduce the likelihood of resistance. The results are consistent with previous studies that emphasize the importance of designing new chemical compounds that target vital bacterial enzymes. Compounds (Q3, Q4, Q6) showed promising potential as novel antibiotics against Proteus mirabilis, with future improvements to improve efficacy and approach that of standard antibiotics.
The results showed a p-value < 0.05, indicating that there were statistically significant differences between the MIC values of the compounds. The compounds (Q3, Q4, Q6) showed clear differences compared to the compounds (Q5, Q7) and the ineffective compound (Q2), with a significantly lower performance compared to Ciprofloxacin. The relationship between chemical structure (number of functional groups, molecular weight) and MIC values was analyzed an inverse correlation between the number of functional groups and MIC values (R² = 0.82, p < 0.05) was found, indicating that increasing the number of active functional groups enhances efficacy. The lower molecular weight compounds (Q3, Q4, Q6) showed greater efficacy compared to the heavier compounds, such as Q5, Q7.
When analyzing the replicates of each compound, the compounds (Q3, Q4, Q6) showed high stability in the results, which enhances the reliability of the efficacy. The T-test between the replicates confirmed that there were no significant differences within the same group for each compound (p > 0.05). Statistical criteria such as the F-statistic in ANOVA and the R² value in the regression showed that the model used to interpret the efficacy based on the chemical structure is strong (R² > 0.8). The compounds (Q3, Q4, Q6) showed high and stable efficacy against Proteus mirabilis compared to the rest of the compounds, which was confirmed statistically. The results show a close correlation between chemical structure and MIC values, with appropriate functional groups enhancing efficacy. The statistical study supports the potential of developing compounds (Q3, Q4, Q6) as promising antibiotics, with a focus on optimizing molecular weight and functional group arrangement to improve efficacy agreed with the study [34].
The study demonstrated the effectiveness of a group of synthetic chemical compounds against Proteus mirabilis bacteria. Compounds Q3, Q4, and Q6 were the most effective, inhibiting bacterial growth with a minimum inhibitory concentration (MIC) of 256 µg/ml, demonstrating their high potential against this type of bacteria. In contrast, compounds Q5 and Q7 showed moderate effectiveness, with an MIC of 512 µg/ml, while compound Q2 showed no antibacterial activity. Comparing these compounds with the standard antibiotic ciprofloxacin, we found that the latter was the most effective, with a MIC of only 2 µg/ml, highlighting its strength as a conventional antibiotic. Although the synthetic compounds did not reach the level of effectiveness of ciprofloxacin, they showed promising potential as alternative antibiotics, with future improvements needed to increase their effectiveness. The chemical structure of compounds plays a crucial role in determining their effectiveness. Compounds containing multiple functional groups, such as (-OH) and (-NH2), were found to be better able to bind to the active site of DNA gyrase, enhancing their antibacterial efficacy. Their low molecular weight and appropriate arrangement of functional groups also contributed to the compounds' enhanced bioactivity. The results of the study indicate that compounds Q3, Q4, and Q6 can be used as prototypes for the development of new antibiotics. However, these compounds require modifications to their chemical structures to enhance their ability to effectively bind to the enzyme and increase their permeability to bacterial cells. The importance of this study lies in highlighting the pivotal role of computational modeling and chemical compound design as an innovative means to combat antibiotic resistance, a growing global health problem. The results also support research efforts aimed at discovering new compounds to combat resistant bacteria such as Proteus mirabilis. Emphasize the potential of the most active compounds (Q3, Q4, Q6) as lead scaffolds, address the need for further structural optimization to enhance bioavailability and potency, and recommend broader biological testing (in vitro and in vivo) to validate safety and efficacy.
The authors would like to thank Mustansiriyah University (www.uomustansiriyah.edu.iq), Baghdad, Iraq, for its support in the present work, and are extremely grateful to the Polymers Research Unit, College of Science, Mustansiriyah University, Baghdad, Iraq, and the Teacher in the Oral and Maxillofacial Surgery Department, College of Dentistry, University of Bilad Alrafidain and Department of Biology, College of Education for Pure Science, Diyala University, Iraq, and University of Diyala, College of science, Department of Forensic Sciences, for their cooperation and all the people help us to get our data.
[1] Livermore, D.M., Blaser, M., Carrs, O., Cassell, (2011). Discovery research: The scientific challenge of finding new antibiotics. Journal of Antimicrobial Chemotherapy, 66(9): 1941-1944. https://doi.org/10.1093/jac/dkr262
[2] Varela, M.F., Stephen, J., Lekshmi, M., Ojha, M., Wenzel, N., Sanford, L.M., Hernandez, A.J., Parvathi, A., Kumar, S.H. (2021). Bacterial resistance to antimicrobial agents. Antibiotics, 10(5): 593. https://doi.org/10.3390/antibiotics10050593
[3] Xu, Z., Xu, D., Zhou, W., Zhang, X. (2022). Therapeutic potential of naturally occurring benzofuran derivatives and hybrids of benzofurans with other pharmacophores as antibacterial agents. Current Topics in Medicinal Chemistry, 22(1): 64-82. https://doi.org/10.2174/1568026621666211122162439
[4] Arandjelovic, P., Doerflinger, M., Pellegrini, M. (2019). Current and emerging therapies to combat persistent intracellular pathogens. Current Opinion in Pharmacology, 48: 33-39. https://doi.org/10.1016/j.coph.2019.03.013
[5] Lande, L., George, J., Plush, T. (2018). Mycobacterium avium complex pulmonary disease. Current Opinion in Infectious Diseases, 31(2): 199-207. https://doi.org/10.1097/qco.0000000000000437
[6] Grosdidier, A., Zoete, V., Michielin, O. (2011). SwissDock, a protein-small molecule docking web service. Nucleic Acids Research, 39(2): W270-W273. https://doi.org/10.1093/nar/gkr366
[7] Yilmaz, S., Yalcin, I., Okten, S., Onurdag, F.K., Aki-Yalcin, E. (2017). Synthesis and investigation of binding interactions of 1, 4-benzoxazine derivatives on topoisomerase IV in Acinetobacter baumannii. SAR and QSAR in Environmental Research, 28(11): 941-956. https://doi.org/10.1080/1062936X.2017.1404490
[8] Ebenezer, O., Singh-Pillay, A., Koorbanally, N.A., Singh, P. (2021). Antibacterial evaluation and molecular docking studies of pyrazole–thiosemicarbazones and their pyrazole–thiazolidinone conjugates. Molecular Diversity, 25(1): 191-204. https://doi.org/10.1007/s11030-020-10046-w
[9] Pacios, O., Blasco, L., Bleriot, I., Fernandez-Garcia, L., (2020). Strategies to combat multidrug-resistant and persistent infectious diseases. Antibiotics, 9(2): 65. https://doi.org/10.3390/antibiotics9020065
[10] Qin, Y., Xu, L., Teng, Y., Wang, Y., Ma, P. (2021). Discovery of novel antibacterial agents: Recent developments in D-alanyl-D-alanine ligase inhibitors. Chemical Biology & Drug Design, 98(3): 305-322. https://doi.org/10.1111/cbdd.13899
[11] Petrella, S., Capton, E., Raynal, B., Giffard, C., Thureau, A., Bonneté, F., Alzari, P.M., Aubry, A., Mayer, C. (2019). Overall structures of Mycobacterium tuberculosis DNA gyrase reveal the role of a corynebacteriales GyrB-specific insert in ATPase activity. Structure, 27(4): 579-589. https://doi.org/10.1016/j.str.2019.01.004
[12] Smith, P.A., Koehler, M.F., Girgis, H.S., Yan, D., et al. (2018). Optimized arylomycins are a new class of Gram-negative antibiotics. Nature, 561(7722): 189-194. https://doi.org/10.1038/s41586-018-0483-6
[13] Newman, D.J., Cragg, G.M. (2016). Natural products as sources of new drugs from 1981 to 2014. Journal of Natural Products, 79(3): 629-661. https://doi.org/10.1021/acs.jnatprod.5b01055
[14] Bush, N.G., Diez-Santos, I., Abbott, L.R., Maxwell, A., (2020). Quinolones: Mechanism, lethality and their contributions to antibiotic resistance. Molecules, 25(23): 5662. https://doi.org/10.3390/molecules25235662
[15] Levin-Reisman, I., Ronin, I., Gefen, O., Braniss, I., Shoresh, N., Balaban, N.Q. (2017). Antibiotic tolerance facilitates the evolution of resistance. Science, 355(6327): 826-830. https://doi.org/10.1126/science.aaj2191
[16] Blower, T.R., Williamson, B.H., Kerns, R.J., Berger, J.M. (2016). Crystal structure and stability of gyrase–fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences, 113(7): 1706-1713. https://doi.org/10.1073/pnas.1525047113
[17] Muteeb, G., Rehman, M.T., Shahwan, M., Aatif, M. (2023). Origin of antibiotics and antibiotic resistance, and their impacts on drug development: A narrative review. Pharmaceuticals, 16(11): 1615. https://doi.org/10.3390/ph16111615
[18] Aragaw, W.W., Cotroneo, N., Stokes, S., Pucci, M., Critchley, I., Gengenbacher, M., Dick, T. (2022). In vitro resistance against DNA gyrase inhibitor SPR719 in Mycobacterium avium and Mycobacterium abscessus. Microbiology Spectrum, 10(1): e01321-21. https://doi.org/10.1128/spectrum.01321-21
[19] Bradford, P.A., Miller, A.A., O'Donnell, J., Mueller, J.P. (2020). Zoliflodacin: An oral spiropyrimidinetrione antibiotic for the treatment of Neisseria gonorrhoeae, including multi-drug-resistant isolates. ACS Infectious Diseases, 6(6): 1332-1345. https://doi.org/10.1021/acsinfecdis.0c00021
[20] Baldwin, S.L., Larsen, S.E., Ordway, D., Cassell, G. (2019). The complexities and challenges of preventing and treating nontuberculous mycobacterial diseases. PLoS Neglected Tropical Diseases, 13(2): e0007083. https://doi.org/10.1371/journal.pntd.0007083
[21] Muhammed, M.T., Aki-Yalcin, E. (2021). Computational insight into the mechanism of action of DNA gyrase inhibitors; revealing a new mechanism. Current Computational Chemistry, 20(3): 224-235. https://doi.org/10.2174/1573409919666230419094700
[22] Muhammed, M.T., Aki-Yalcin, E. (2021). Pharmacophore modeling in drug discovery: Methodology and current status. Journal of the Turkish Chemical Society, Section A: Chemistry, 8(3): 749-762. https://doi.org/10.18596/jotcsa.927426
[23] Qidwai, T. (2017). QSAR modeling, docking and ADMET studies for exploration of potential anti-malarial compounds against Plasmodium falciparum. In Silico Pharmacology, 5(1): 6. https://doi.org/10.1007/s40203-017-0026-0
[24] Waterhouse, A., Bertoni, M., Bienert, S., Studer, (2018). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1): W296-W303. https://doi.org/10.1093/nar/gky427
[25] Latif, A.S., Magtooph, M.G., Alubadi, A.E.M. (2020). In silico and In vitro evaluation of some synthesized quinoline derivatives into MexB protein of Pseudomonas aeruginosa. International Journal of Drug Delivery Technology, 10(2): 195-199.
[26] Dahlgren, D., Lennernäs, H. (2019). Intestinal permeability and drug absorption: Predictive experimental, computational and in vivo approaches. Pharmaceutics, 11(8): 411. https://doi.org/10.3390/pharmaceutics11080411
[27] Martin, Y.C. (2005). A bioavailability score. Journal of Medicinal Chemistry, 48(9): 3164-3170. https://doi.org/10.1021/jm0492002
[28] Muhammed, M.T., Kuyucuklu, G., Kaynak-Onurdag, F., Aki-Yalcin, E. (2022). Synthesis, antimicrobial activity, and molecular modeling studies of some benzoxazole derivatives. Letters in Drug Design & Discovery, 19(8): 757-768. https://doi.org/10.2174/1570180819666220408133643
[29] Gordeev, M.F., Yuan, Z.Y. (2014). New potent antibacterial oxazolidinone (MRX-I) with an improved class safety profile. Journal of Medicinal Chemistry, 57(11): 4487-4497. https://doi.org/10.1021/jm401931e
[30] Christensen, S.B. (2021). Drugs that changed society: History and current status of the early antibiotics: Salvarsan, sulfonamides, and β-lactams. Molecules, 26(19): 6057. https://doi.org/10.3390/molecules26196057
[31] Tian, W., Chen, C., Lei, X., Zhao, J., Liang, J. (2018). CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Research, 46(W1): W363-W367. https://doi.org/10.1093/nar/gky473
[32] Taylor, S.N., Marrazzo, J., Batteiger, B.E., Hook, E.W., et al. (2018). Single-dose zoliflodacin (ETX0914) for treatment of urogenital gonorrhea. The New England Journal of Medicine, 379: 1835-1845. https://doi.org/10.1056/NEJMoa1706988
[33] Basarab, G.S., Kern, G.H., McNulty, J., Mueller, J.P (2015). Responding to the challenge of untreatable gonorrhea: ETX0914, a first-in-class agent with a distinct mechanism-of-action against bacterial Type II topoisomerases. Scientific Reports, 5(1): 11827. https://doi.org/10.1038/srep11827
[34] Latif, A.S., Fadel, Z.A. (2022). Evaluation study of the effectiveness for some antibacterial agent against dna gyrase enzyme of Staphylococcus aureus. European Chemical Bulletin, 11(7): 29-32.