Electrochemical Performance of Cupric Oxide Loaded Carbon Nanotubes as Electrode Material for CO2 Reduction

Sami M. Ibn Shamsah*
© 2021 IIETA. This article is published by IIETA and is licensed under the CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
OPEN ACCESS
This study is conducted to explore the effective and stable electrode materials for electrochemical reduction of CO2. The different compositions (3 - 8%) CuO-supported CNT samples were prepared by a traditional sol-gel method, and the prepared materials were characterized by TGA, SEM, TEM, FTIR and XRD. The characterization results confirmed the uniform impregnation of CuO on the defects of the CNT's, which improved the utilization of meso and microporous distribution in the prepared electrocatalyst. The high surface area and stability of CuO- CNTs allowed dual conduction of electron and intermediate species with high current density at minimum energy supplied. Linear sweep voltammetry and Chronoamperometry were used to measure the electrode's current density and stability, respectively, and 8 % CuO- CNT was found most stable and progressive composition to reduce the CO2 efficiently. Further, the liquid products were analyzed using the gas chromatography, and 20 % faradic efficiency of methane was measured.
carbon nanotubes, cupric oxide, CO2 reduction, electrochemical cell, linear sweep voltammetry
Extreme dependence on fossil fuel has raised the CO2 concentration in the atmosphere and underscored the reservations on fossil fuel dependence in the future. A need for sufficient and clean energy resources is crucial to control the CO2 emissions [1, 2]. Many efforts have been carried out to transform fossil fuel to renewable energy sources to meet social, environmental, and economic targets set in the Kyoto protocol [3]. The conversion of CO2 is one of the most useful ways to utilize renewables energy to convert the CO2 into valuable hydrocarbons and alcohol [4, 5].
Carbon dioxide is being reduced to fuel on an industrial scale using the hydrogenation process. The high temperature and pressure requirement with a considerable quantity of hydrogen put some limitations on the industrial process [6]. A series of photocatalysts has also been used for the photochemical reduction of CO2 to alcohol. The insufficient light intensity and electron movement back to the hole restrict photochemical catalysts [7, 8]. Electrochemical reduction of CO2 is an essential route for converting CO2 to valuable products at ambient temperature and pressure. Different systems (aqueous and non-aqueous) have been used to produce hydrocarbons and alcohols, such as methanol, ethylene, methane, and ethanol [4, 9, 10].
Before the1980, CO2 has been reduced to formic acid over zinc and mercury metal with a very low selectivity [11]. In the 1980s, Frese et al. were the first to reduce CO2 into the most desirable product (Methanol) but with a very low current density [12, 13]. Later, Hori et al. obtained CO and hydrogen evolutions as a primary reduction product when used a series of different metals Ni, Pd, Au, Zn, Ag, Pt, Fe, Rh, Mo, and Ti. Hori et al. continued to develop the novel materials and found Copper to be the most promising material to reduce CO2 into CH4 and ethylene with a limited selectivity [14-17].
Since the excellent work by Hori et al. [18]., many researchers have worked on tuning and controlling the structure of Copper-based electrocatalysis to demonstrate better efficiency, high selectivity, and achieving good accessibility of electrons on the cathode surface [19-25].
The main hurdle in the electrochemical CO2 reduction process is the reduction of water at the cathode surface. The reduction potential of water is very close to the reduction potential of CO2, and a small window of CO2 potential allows water to consume electron at the cathode surface, which eventually reduces the selectivity of the product. Similarly, low current density and low overpotential for H2O reduction are the significant problems faced in the electrochemical reduction of CO2. In addition to that, few electrons' accessibilities on the electrode's surface and less participation of electrons in the conduction band are the significant obstacles faced in the process [26-30].
Therefore, a metal-based electrocatalyst with meso and microporous support to increase the adsorbents' residence time are very convenient. It has been studied that the tubular structure of carbon nanotubes enables the pores to behave as molecular wires for electron transport between the electrode and electrolyte solution and eventually increases the electron's availability on the cathode surface [31-33].
In this work, functionalized carbon nanotubes (CNTs) impregnated with 5 to 8 % Cupric oxide (CuO) were investigated as a cathode material for the electrochemical reduction of CO2. The surface and structural properties of electrocatalyst (CuO-CNTs) with different loadings were characterized using various techniques, including SEM, TEM, XRD and TGA. The Linear sweep voltammetry and a chronoamperometry test were performed to measure the stability and correlate the electrochemical properties with the electrocatalyst.
2.1 Preparation of CuO (3,5 and 8 %) supported CNTs
The synthesis of Cupric oxide (CuO) supported carbon nanotubes (CNTs) has been carried out by the traditional Sol-gel method. Initially, a mixture of CNTs and ethanol was sonicated for thirty minutes with an amplitude of 50% in the full frequency range. Later the desired weight percent of copper nitrate pentahydrate salt was introduced in CNTs ethanol slurry, and the mixture was further sonicated for thirty-five minutes with the same frequency to ensure the fair distribution of copper salt over CNTs. The addition of copper salt in the slurry depends upon wt.% of CuO needed in the end product, i.e., 3% - 8%. After further sonication, the mixture was placed in an oven at a temperature of 85 oC for 24 hours, and later, the dried cake was calcined in the furnace at 360 oC for 3 to 4 hours. The prepared calcined product was cooled to room temperature, and CuO supported CNTs electrocatalysts were obtained and stored in the bag.
2.2 Carbon nanotubes (CNTs) functionalization
The CNTs were functionalized to attach the COOH group and create the surface's defects. 2 grams of CNTs were sonicated for thirty-five minutes in 90 ml of concentrated HNO3 and H2SO4 solution. After that, the sonicated mixture was vigorously stirred and refluxed in a round bottle flask for 48 hours at 120oC. The mixture was cooled, washed with deionized water, and centrifuged at 15,000 rpm. Later the functionalized CNTs were placed in a vacuum oven at 85 °C for ten hours.
2.3 Experimental setup
Two compartments Pyrex glass electrochemical cell with reference, counter, and working electrodes were used to record the Linear sweep voltammetry (LSV) and chronoamperometry measurements. For each batch of the experiment, CuO supported CNTs were sonicated with Nafion and isopropanol solution; after that, the slurry was placed on copper foil and allowed to dry and stuck on the foil surface. Before the start of each experiment. Nitrogen was bubbled in the cell to remove the air from the compartment and protect the working electrode surface from the air's oxygen contents.
3.1 Characterization of Functionalized Carbon nanotubes (CNTs.)
3.1.1 FTIR Analysis
FTIR analysis was used to indicate the presence of the carboxylic group in the functionalized CNTs. Figure 1 represents the FTIR analysis of the functionalized CNTs. The carbonyl stretching in the COOH group was observed at 1741 cm−1. Peaked at 3429 cm−1, indicating OH groups' presence while phenyl-carbonyl C–C stretch was noticed at 1382 cm−1. The appearance of carboxylic groups in the functionalized CNT has made the material more suitable to attach CuO with the CNT's defects.
Figure 1. FTIR spectra of Functionalized CNTs
3.2 Characterization CuO supported CNTs
3.2.1 Thermal gravimetric analysis (TGA)
Thermogravimetric analysis (TGA) was carried out to record moisture or volatile material present in the sample and explored the material's thermal strength. TGA experiment was performed under the temperature range of 25 to 600oC using the alumina pan and air purging system with 100 ml/minute. Figure 2 shows the TGA curves recorded for 3 to 8 % CuO supported CNTs and indicates carbon oxidation in the temperature range of 420 to 480oC. The carbon burning has caused a decline in weight loss for each sample, and 3% CuO supported CNTs had the highest weight drop than higher loaded materials because of a higher percentage of carbon present in the sample. Higher loaded CuO materials went through early oxidation around 430oC, showing that higher loaded CuO materials are thermally weak but have better encapsulation than less impregnated CuO based materials.
Figure 2. TGA curves of 3, 5, and 8 % CuO supported CNTs
3.2.2 XRD (X-ray powder diffraction)
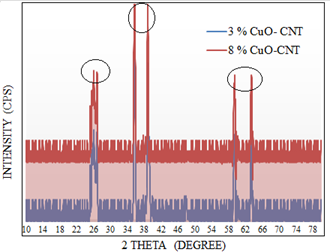
XRD analysis was carried out to ensure the crystal size and phase analysis. Figure 3 shows the XRD analysis of CNTs- supported CuO electrocatalysts ranges from 3,5 and 8%. The diffraction peak at 26 o represents hexagonal graphite (002) planes, while peaks at 36.3o, 38.2o, 59o and 63o indicate divalent and monoclinic nanocrystallites of CuO, which confirmed the presence of CuO supported CNTs and validates the method of preparation. The higher intense peaks were observed for high loaded materials (8 % CuO-CNT), suggesting that highly impregnated materials exhibit a strong intensity. The mean particle size of 3,5 and 8% CuO–CNTs was calculated using Scherrer's equation based on the strongest diffraction peak corresponding to CuO (111)
Scherrer's equation:
$D=0.9 \lambda / \beta \operatorname{Cos} \theta$
where λ is the X-ray wavelength, β is the FWHM of the peak, and θ is the diffraction angle.
Table 1. The crystallite size of (3 and 8 %) CuO supported CNTs
|
Electrocatalyst Sample |
Crystallite size |
|
3 % CuO -CNTs |
3.35 nm |
|
5 % CuO -CNTs |
4.4 nm |
|
8 % CuO -CNTs |
6.0 nm |
Figure 3. XRD patterns of (3 and 8 %) CuO supported CNTs
3.2.3 Scanning Electron Microscopy (SEM) and Energy-dispersive X-ray spectroscopy (EDX)
Figure 4. SEM images of 3% CuO supported CNTs (a) 5% CuO supported CNTs (b) and 8% CuO supported CNTs (c)
The SEM analysis was carried out to study the surface morphology and ensure the materials' homogenous distribution. Figure 4 shows the SEM images of 3, 5, and 8 % CuO- CNT samples. The tubular geometry of CNTs in fig 5 confirmed that the tubes' geometry has not changed after the synthesis. Homogeneous coverage of CuO-CNT is visible in fig 5 for 3, 5, and 8 % sample indicates the uniformity and effectiveness of the synthesis process. However, higher loaded CuO – CNTs, e.g., 8 % formed agglomerates, are visible in fig 4 (c).
3.2.4 Transmission Electron Microscopy (TEM)
The TEM images of 3 and 8% CuO- CNTs electrocatalysts are given in Figure 5. The TEM test was conducted to calculate the tube's diameter and the crystal size of the materials. The size of 3% CuO-CNTs particle is 3.4 nm measured by TEM has matched with XRD analysis, and the diameter of tubes was recorded around 4-11 nm that was in agreement with the manufacturer specification and confirmed that impregnation of the CuO on CNT had not changed the geometry of the CNTs materials, the uniform attachment of the CuO over the CNTs is visible in Figure 5.
Figure 5. TEM images of 3% CuO supported CNTs (a), 8% CuO supported CNTs (b)
3.2.5 Brunauer Emmett Teller (BET)
BET isotherms for each sample tabulated in table 2 were matched with the typical type 3 adsorption model as per the IUPAC system; the pore size distribution was uniformed despite the CuO’s attachment with the CNTs. The BET analysis confirmed the decline in surface area for higher loaded particles.
Table 2. BET Surface area of (3, 5 and 8 %) CuO supported CNTs
|
Electrocatalyst |
BET Surface area (m2/g) |
|
CNTs |
165 |
|
3% CuO-CNT |
160 |
|
5% CuO-CNT |
156 |
|
8% CuO-CNT |
154 |
3.3 Linear sweep Voltammetry
The linear sweep voltammetry (LSV) test was recorded in a three-compartment electrochemical cell separated by the Nafion membrane; the reference, counter, and working electrodes were used to evaluate the performance of the catalyst. Before start of each experiment, N2 was introduced with a high pressure in the cell to remove the air contents present in the compartment, and later the CO2 was bubbled for thirty-five minutes in a 0.5 M KaHCO3 electrolyte solution. The electrocatalyst (3-8% CuO-CNTs) slurry was prepared with Nafion and isopropanol solution, and the catalyst ink was introduced on the copper foil and allowed to dry. LSV analysis was carried out to evaluate the catalyst's electrochemical performance in the potential range of -0.3 to -1.7 V. Figure 6 represents the response of each electrocatalyst against the potential applied. A rise in current density was observed from the onset of the potential, and it was continuously increasing within the entire potential window. However, 8% loaded CuO - CNT had the highest current density than the rest of the materials, the rise in current density for higher loaded (3-8% CuO-CNT) materials was due to large particle size uniform impregnation of CuO particles over the CNTs surfaces observed during XRD, TEM, and SEM analysis. The growth in particle size and less surface area of higher loaded materials, as confirmed form BET analysis is a better tradeoff between particle size and catalytic activity which promoted the electron flow and its availability for CO2. The 8% CuO-CNT sample had the optimum crystal size to reduce the CO2 efficiently. The suitable crystal size of the 8% CuO-CNT ensures the timely availability of adsorbed species required to reduce CO2 into most desirable products.
Figure 6. Linear Sweep Voltammetry (LSV) curves of copper foil with CO2 saturated electrolyte and Copper foil coated with (3,5 and 8%) CuO- supported CNTs
3.4 Chronoamperometry Experiments
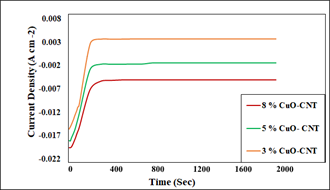
Figure 7. Chronoamperometry curves of Copper foil coated with (3,5 and 8%) CuO- supported CNTs
Figure 7 shows the Chronoamperometry test recorded for 1900 seconds to measure the electrocatalyst's stability at the constant potential of -1.5 V. Variation of current density for different loaded CuO -CNT materials is visible in figure 7. The sudden rise in current density for each sample until the 200 seconds was recorded; however, after 200 seconds, a constant profile was achieved. The loading of CuO on the CNT surface has increased the current density, and the highest current density (-0.020 A cm-2) was observed at -1.5 V for 8% CuO- CNT electrocatalyst, which agrees with the LSV results. Higher loaded materials, e.g., 8% CuO-CNT, significantly increased the current density because of the good agreement between surface area and crystal size. The increased pores size distribution in highly impregnated materials ultimately increased a current density. Similarly, no side reaction was observed during the experiment, ensured the catalyst's stability for 1900 seconds.
3.5 Faradic efficiency
A Chronoamperometry experiment was performed for 1900 seconds at each constant potential value from -0.3 to -1.7 V under 8 % CuO-CNTs sample to analyze the liquid products. The liquid products were analyzed in a GC equipped FID detector under the standard internal method, and faradic efficiency was calculated based on the following equation.
$\frac{f \times m \times n}{1 \times t}$
F is faraday’s constant, m is the number of moles of ethanol formation; I is total current, t is in seconds, and n is the number of electrons involved in methane formation. The 20 % faradic efficiency of methane observed at a potential of -1.5 V.
CuO-supported functionalized CNTs were prepared with different loadings, and prepared materials were tested for the electrochemical reduction of CO2. The characterization and electrochemical results showed that CuO-CNTs had an excellent performance to convert the CO2 into the methane at the minimum potential range. Loading of CuO particles on the CNTs exhibited an excellent current density, and 8% CuO-CNTs was recorded as a highly efficient and stable electrocatalyst to adsorbed the CO2 on the meso- and microporous surface. A 8% CuO-CNT provided the intermediate species' a timely availability of electrons because tubular tubes in the CNTs behaved as electron wire. The firmed attachment of CuO on the defected side of CNTs, as confirmed from FTIR, enhanced the electron flow on the cathode surface to converts CO2 at the minimum potential value. Higher loading CuO-supported CNTs had large crystalline size, better pore size distribution, and its optimum attachment with defects of CNT showed high current density, and better stability, as confirmed by BET, SEM. TEM, XRD, and chronoamperometry. Adequate metal oxide loading and a trade-off between particle size and active sites are essential to reduce CO2 efficiently.
The author would like to acknowledge the support provided by the University of Hafr Al Batin, Kingdom of Saudi Arabia.
Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
[1] Kim, Y., Tanaka, K., Matsuoka, S., Environmental and economic effectiveness of the Kyoto Protocol. Plos one, 15(7): e0236299 (2020). https://doi.org/10.1371/journal.pone.0236299
[2] Abas, N., Kalair, A., Khan, N., Futures, 69, 31 (2015). https://doi.org/10.1016/j.futures.2015.03.003
[3] Wang, C.H., Ko, M.H., Chen, W.J., Sustainability, 11(3), 744 (2019). https://doi.org/10.3390/su11030744
[4] Lee, M.Y., Park, K.T., Lee, W., Lim, H., Kwon, Y., Kang, S., Critical Reviews in Environmental Science and Technology, 50(8), 769 (2020). https://doi.org/10.1080/10643389.2019.1631991
[5] Al-Rowaili, F.N., Jamal, A., Ba Shammakh, M.S., Rana, A., ACS Sustainable Chemistry & Engineering, 6(12), 15895 (2018). https://doi.org/10.1021/acssuschemeng.8b03843
[6] Marlin, D.S., Sarron, E., Sigurbjörnsson, Ó., Frontiers in Chemistry, 6, 446 (2018). https://doi.org/10.3389/fchem.2018.00446
[7] Wu, J., Huang, Y., Ye, W., Li, Y., Advanced Science, 4 (11), 1700194 (2017). https://doi.org/10.1002/advs.201700194
[8] Kumar, B., Llorente, M., Froehlich, J., Dang, T., Sathrum, A., Kubiak, C.P., Annual review of Physical Chemistry, 63, 541 (2012). https://doi.org/ 10.1146/annurev-physchem-032511-143759
[9] Nitopi, S., Bertheussen, E., Scott, S.B., Liu, X., Engstfeld, A.K., Horch, S., Chorkendorff, I., Chemical Reviews, 119 (12), 7610 (2019). https://doi.org/10.1021/acs.chemrev.8b00705
[10] Chen, C., Kotyk, J.F.K., Sheehan, S.W., Chem, 4 (11), 2571 (2018). https://doi.org/10.1016/j.chempr.2018.08.019
[11] Paik, W., Andersen, T.N., Eyring, H., Electrochimica Acta, 14 (12), 1217 (1969). https://doi.org/10.1016/0013-4686(69)87019-2
[12] Frese Jr, K.W., Canfield, D., Journal of the Electrochemical Society, 131 (11), 2518 (1984). https://doi.org/10.1149/1.2115351
[13] Kim, J.J., Summers, D.P., Frese Jr, K.W., Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 245(1-2), 223 (1988). https://doi.org/10.1016/0022-0728(88)80071-8
[14] Hori, Y., Kikuchi, K., Suzuki, S., Chemistry Letters, 14(11), 1695 (1985). https://doi.org/10.1246/cl.1985.1695
[15] Hori, Y., Kikuchi, K., Murata, A., Suzuki, S., Chemistry Letters, 15(6), 897 (1986). https://doi.org/10.1246/cl.1986.897
[16] Hori, Y., Koga, O., Yamazaki, H., Matsuo, T., Electrochimica Acta, 40(16), 2617 (1995). https://doi.org/10.1016/0013-4686(95)00239-B
[17] Hori, Y., Takahashi, R., Yoshinami, Y., Murata, A., The Journal of Physical Chemistry B, 101(36), 7075 (1997). https://doi.org/10.1021/jp970284i
[18] Hori, Y., Murata, A., Takahashi, R., Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases, 85(8), 2309 (1989). https://doi.org/10.1039/F19898502309
[19] Zheng, Y., Vasileff, A., Zhou, X., Jiao, Y., Jaroniec, M., Qiao, S.Z., Journal of the American Chemical Society, 141(19), 7646 (2019). https://doi.org/10.1021/jacs.9b02124
[20] Ajmal, S., Yang, Y., Tahir, M.A., Li, K., Nabi, I., Liu, Y., Zhang, L., Catalysis Science & Technology, 10(14), 4562 (2020). https://doi.org/10.1039/d0cy00487a
[21] Popović, S., Smiljanić, M., Jovanovič, P., Vavra, J., Buonsanti, R., Hodnik, N., Angewandte Chemie International Edition, 59(35), 14736 (2020). https://doi.org/10.1002/anie.202000617
[22] Zhao, J., Xue, S., Barber, J., Zhou, Y., Meng, J., Ke, X., Journal of Materials Chemistry A, 8(9), 4700 (2020). https://doi.org/10.1039/c9ta11778d
[23] Zhang, J., Luo, W., Züttel, A., Journal of Materials Chemistry A, 7(46), 26285 (2019). https://doi.org/10.1039/c9ta06736a
[24] Zhong, S., Electrochemical CO2 Reduction to Value-added Chemicals on Copper-based Catalysts (Doctoral dissertation) (2019).
[25] Vasileff, A., Zhi, X., Xu, C., Ge, L., Jiao, Y., Zheng, Y., Qiao, S.Z., ACS Catalysis, 9(10), 9411 (2019). https://doi.org/10.1021/acscatal.9b02312
[26] Costentin, C., Robert, M., Savéant, J. M., Chemical Society Reviews, 42(6), 2423 (2013). https://doi.org/10.1039/c2cs35360a
[27] Zhu, D.D., Liu, J.L., Qiao, S.Z., Advanced Materials, 28(18), 3423 (2016). https://doi.org/10.1002/adma.201504766
[28] Bailey, S., Froment, G.F., Snoeck, J.W., Waugh, K.C., Catalysis Letters, 30(1), 99 (1994). https://doi.org/10.1007/BF00813676
[29] Malik, M.I., Malaibari, Z.O., Atieh, M., Abussaud, B., Chemical Engineering Science, 152, 468 (2016). https://doi.org/10.1016/j.ces.2016.06.035
[30] Malik, M.I., Electrochemical Reduction of CO2 - ProQuest, King Fahd University of Petroleum and Minerals (2015).
[31] Dumitrescu, I., Unwin, P.R., Macpherson, J.V., Chemical Communications, 2009(45), 6886 (2009). https://doi.org/10.1039/b909734a
[32] Pumera, M., Chemistry–A European Journal, 15(20), 4970 (2009). https://doi.org/10.1002/chem.200900421
[33] Vashist, S.K., Zheng, D., Al-Rubeaan, K., Luong, J.H., Sheu, F.S., Biotechnology Advances, 29(2), 169 (2011). https://doi.org/10.1016/j.biotechadv.2010.10.002